Download to read offline















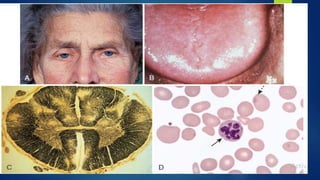




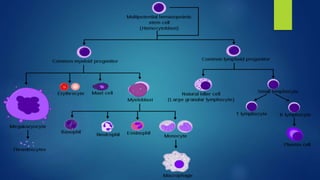
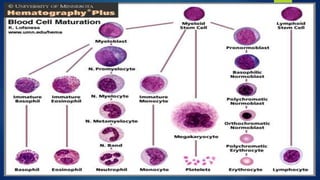



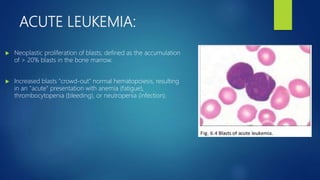
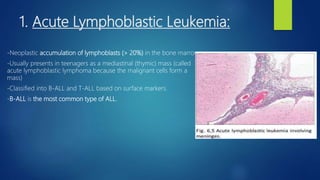



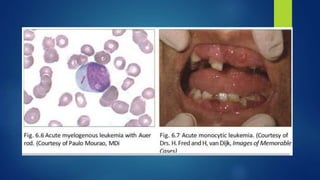


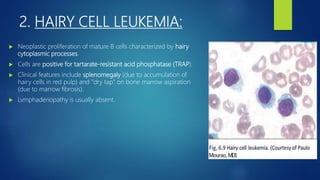


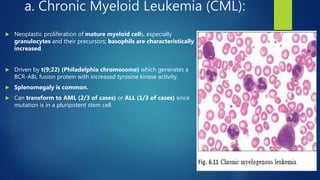

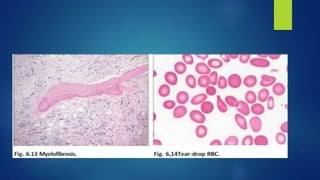










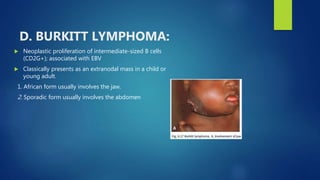








This document provides an overview of several topics in hematology, including: - Anemia, which is a reduction in red blood cells. Common types are discussed such as iron deficiency anemia and megaloblastic anemia. - Leukemia, which are cancers of the white blood cells. The main types - acute and chronic leukemias - are defined. Acute leukemias like ALL and AML are discussed in more detail. - Other blood disorders like lymphomas and lymphadenopathy are listed as learning objectives but not described further in this document.


























![64965 hemodyn[1] edema](https://cdn.slidesharecdn.com/ss_thumbnails/64965hemodyn1-edema-150608143733-lva1-app6892-thumbnail.jpg?width=640&height=640&fit=bounds)




















![BLOOD_DISORDERS-2[1].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/blooddisorders-21-230610142333-f4472c75-thumbnail.jpg?width=640&height=640&fit=bounds)













![Hypothalamus short ppt by Dr. Neha [PT].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/hypothalamusbydr-260124145759-b9f94a93-thumbnail.jpg?width=640&height=640&fit=bounds)














