Downloaded 22 times

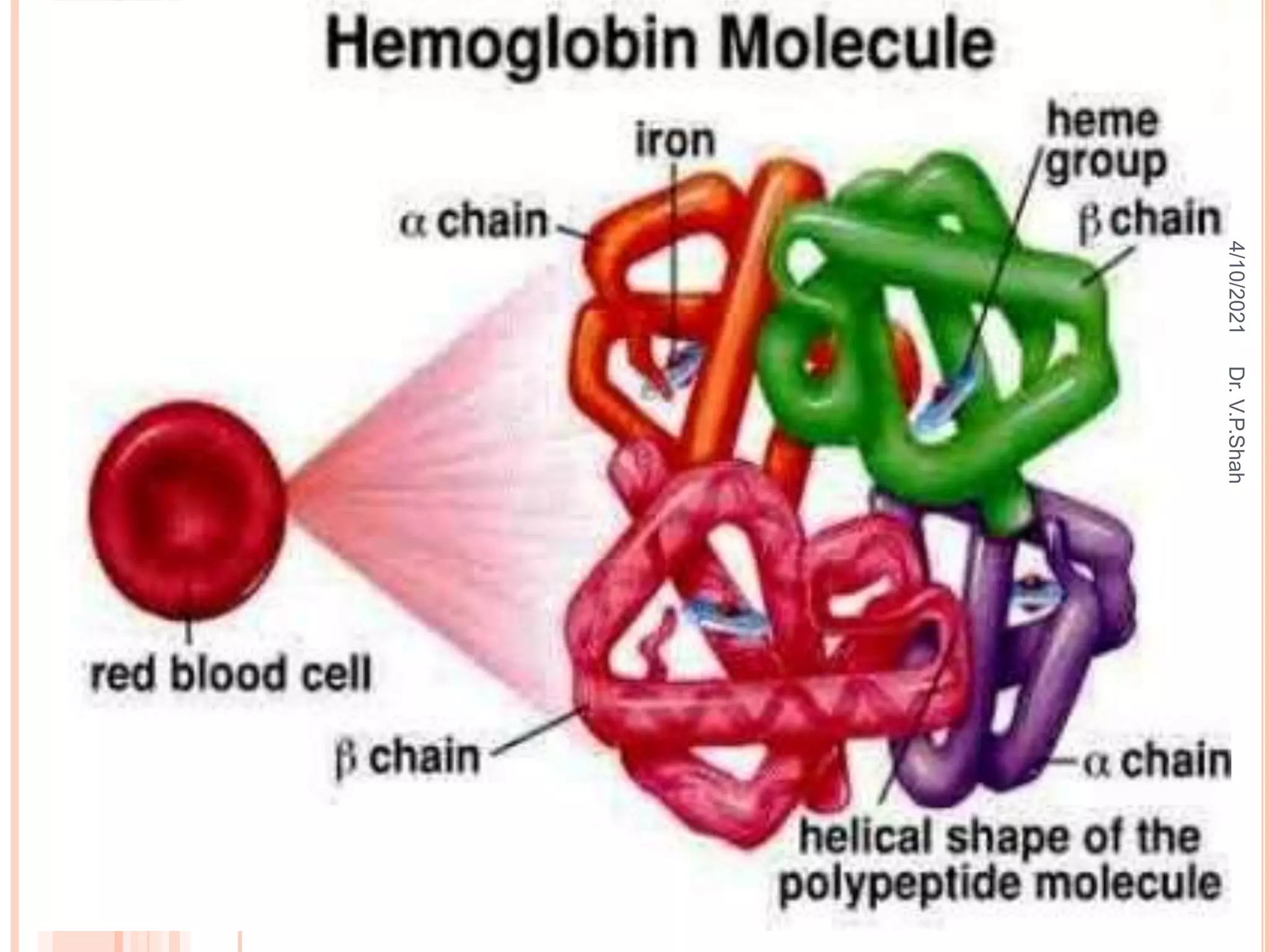

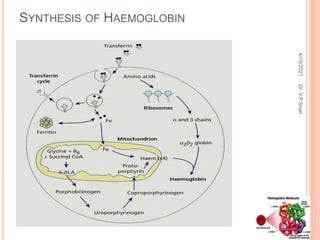
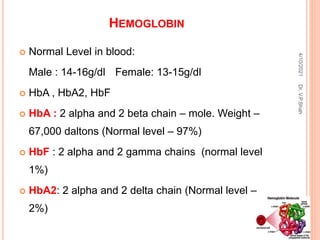

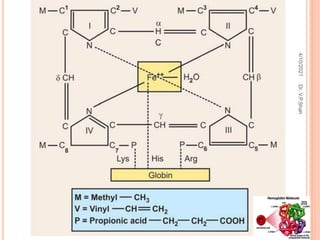



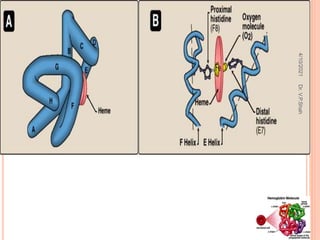

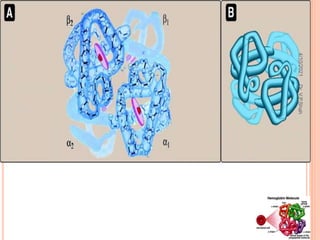
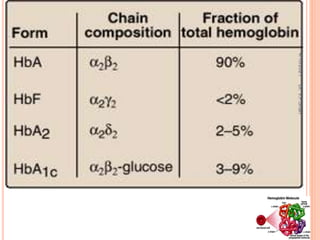



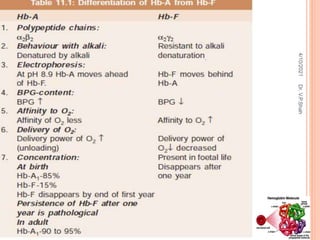

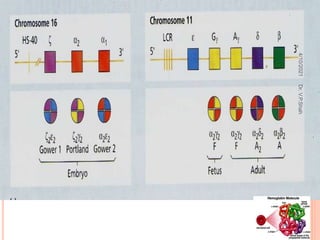
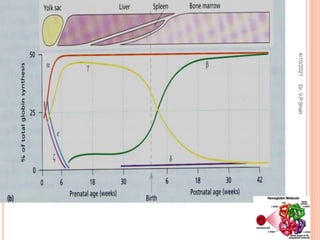

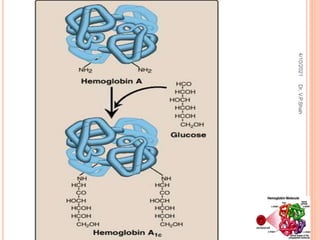

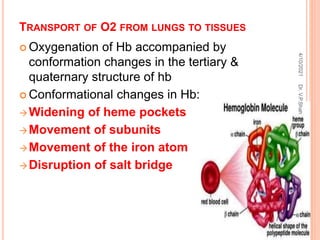
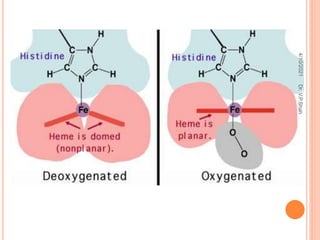

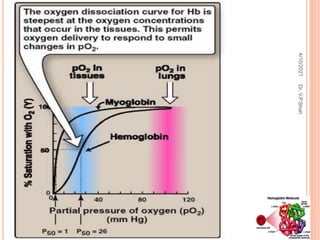

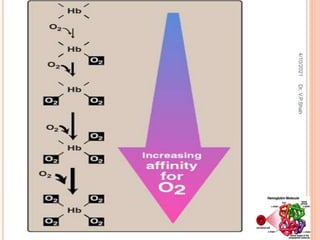


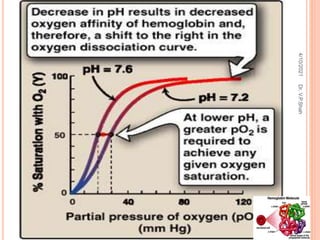

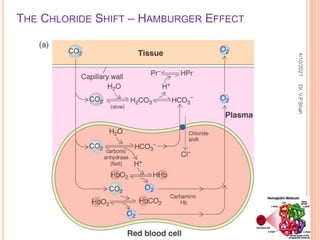
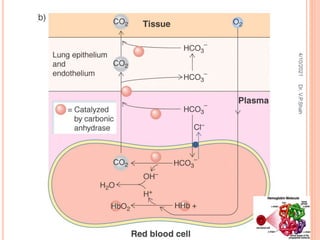

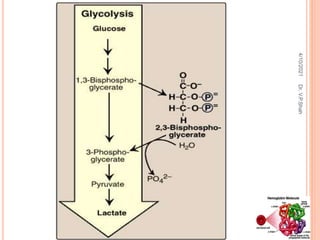
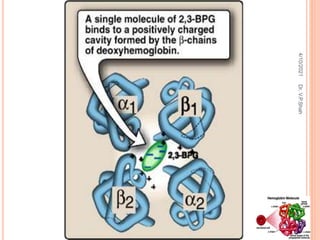
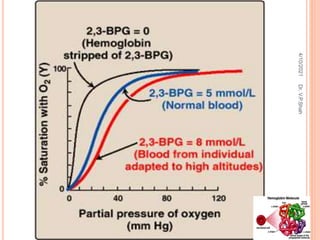

















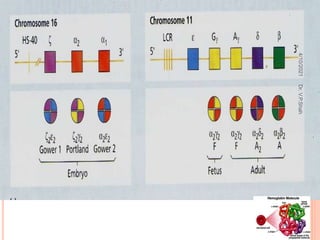

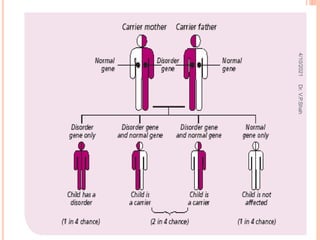
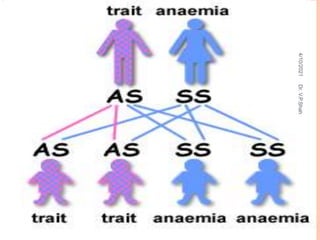






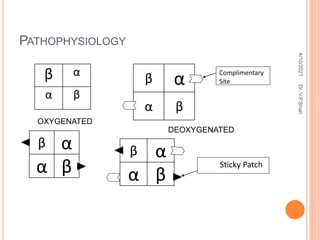
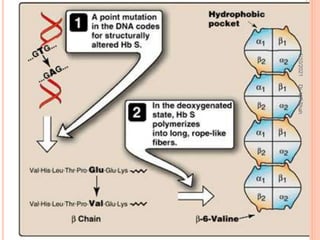

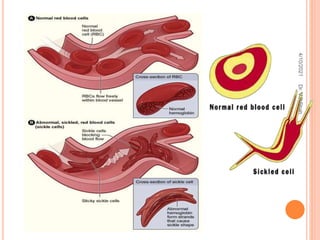

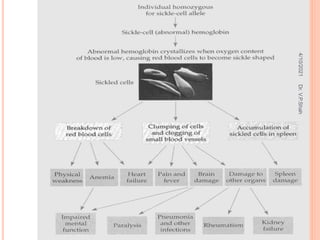


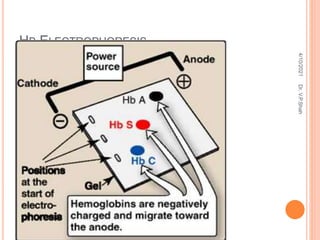






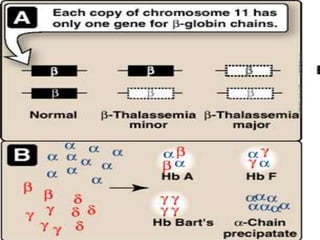





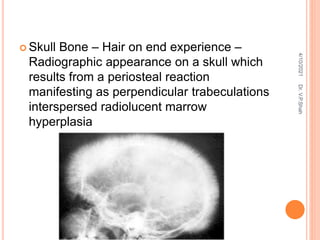






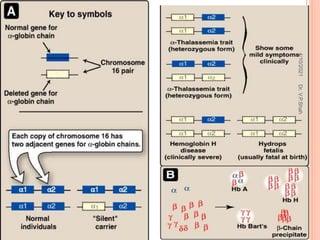













The document discusses the history and structure of hemoglobin. It describes key discoveries such as the identification of red blood cells in 1665, the isolation of hemoglobin in 1862, and the determination of hemoglobin's role in oxygen transport in 1904. The document then provides details on the structure of hemoglobin, including that it is composed of heme and globin. Hemoglobin contains four heme groups, each containing an iron ion, and it exists as an alpha-2 beta-2 tetramer in its main form HbA. The document also reviews factors that affect hemoglobin's oxygen binding such as pH, temperature, and 2,3-BPG levels.



































































