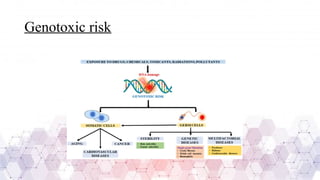
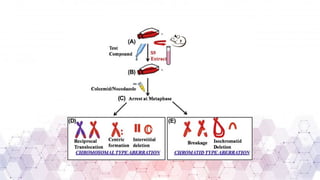
Genotoxicity refers to the ability of chemical, physical, or biological agents to damage genetic material, leading to mutations, chromosomal aberrations, or DNA strand breaks. These genetic alterations can result in various adverse effects, including cancer, reproductive toxicity, and hereditary diseases. Genotoxic substances interact with DNA directly or indirectly, affecting cellular functions and integrity.
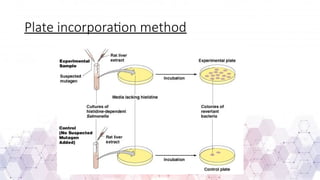
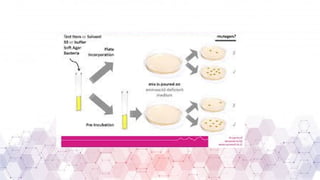
Genotoxicity testing is crucial in pharmaceuticals, environmental sciences, and regulatory assessments to identify potential risks associated with new drugs, chemicals, or pollutants. Common testing methods include the Ames test, micronucleus assay, and comet assay, which help evaluate DNA damage and mutation potential.
Understanding genotoxicity is essential for developing safer compounds and ensuring public health protection by minimizing exposure to harmful agents.