Downloaded 24 times


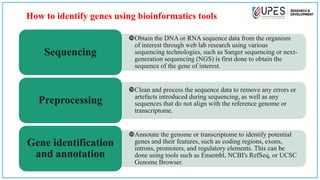

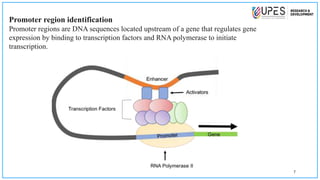


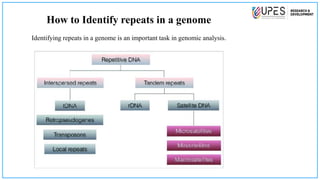
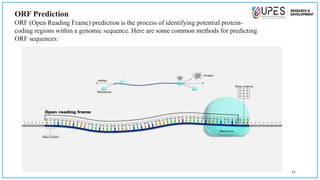




This document discusses various bioinformatics tools and methods for identifying genes from genomic sequences. It begins by defining genes and genomes, then describes reference databases like RefSeq that are important for gene identification. It outlines the general workflow for gene identification, including obtaining sequences, preprocessing, annotation, prediction, and validation. Specific tools mentioned include GENSCAN, Glimmer, and Augustus for gene prediction, and BLAST for sequence alignment. The document also discusses identifying other genomic features like promoters, repeats, and open reading frames. It emphasizes that accurate gene identification requires both computational and experimental approaches.



















































































