
Download as PDF, PPTX














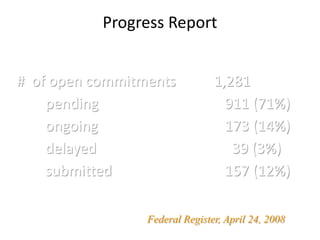



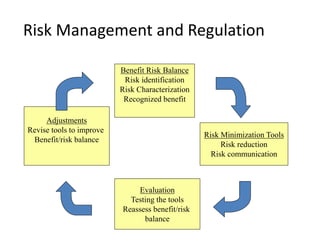



The document discusses the FDA's post-marketing study commitments and the evaluation of drug safety, highlighting the significant public health burden from serious adverse drug events that result in approximately 100,000 deaths annually. It emphasizes the need for improved pre-approval and post-marketing safety evaluations, including more rigorous trials and proactive monitoring of safety signals. The document also underscores the challenges faced by the FDA concerning compliance with safety commitments, approval processes, and the necessity for better enforcement tools to ensure drug safety.




































































![PERI-PROSTHETIC FRACTURE NAIL-PLATE CONSTRUCT [NPC].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/drarunkumardrmohamedashrafperiprostheticfrasturenail-plateconstructnpc-260209164459-7e9d15a1-thumbnail.jpg?width=640&height=640&fit=bounds)


















