This document discusses renal and hepatic encephalopathy. It defines uremic encephalopathy as a brain disorder that occurs in patients with untreated or inadequately treated kidney disease. Symptoms range from mild to severe and fluctuate depending on kidney function. Uremic toxins that accumulate due to renal dysfunction are a primary cause. Treatment involves optimizing dialysis to adequately remove toxins. Other types of encephalopathy discussed include dialysis dementia, central pontine myelinolysis, seizures, and restless leg syndrome. Causes, clinical features, diagnosis and management are described for each condition.












![Dialysis dementia
• Also known as progressive myoclonic dialysis encephalopathy, dialysis
encephalopathy or haemodialysis encephalopathy .
• prevalence -0.6-1.0%.
• time of onset of symptoms had been found to vary from early as in the first month to
several years [upto 9 yr { mean of 3.5 yr}] after dialysis .
• the early and commonest (occurring in 95%) manifestations of dialysis dementia are
speech problems (mixed dysarthria and dysphasia with dysgraphia), apathy and
depression.
• Severe forms have rapid and persistent deterioration of speech, myoclonus (in up to
80%), ataxia and apraxia. In late stages, there are seizures, psychosis
(hallucinations and paranoid delusions) (in up to 60%) and frank dementia in 95% of
cases. In very late stages, patients become immobile and mute.](https://image.slidesharecdn.com/encepalopathy-201122080338/85/Encepalopathy-14-320.jpg)
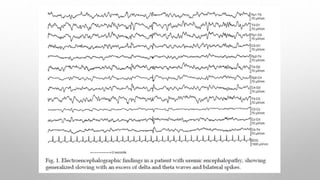
![In untreated cases, death usually occurs within 6-9 months. EEG picture is
similar to that seen in uremic encephalopathy.
Treatment of dialysis dementia
• Dialysis dementia is treated by using desferrioxamine. It is a chelating
agent which binds aluminum with
Greater affinity than that of the plasma proteins [desferrioxaminealuminium
complex].
• This results in improvement of up to 70% of cases, however the clinical
improvement is slow and therapy may need to be given once weekly for
over a year](https://image.slidesharecdn.com/encepalopathy-201122080338/85/Encepalopathy-15-320.jpg)









































































![MANAGEMENT OF METABOLIC ENCEPHALOPATHY [Autosaved].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/managementofmetabolicencephalopathyautosaved-250804190212-5d351afe-thumbnail.jpg?width=640&height=640&fit=bounds)














![Hypothalamus short notes on location, function and disorders by Dr. Neha [PT]...](https://cdn.slidesharecdn.com/ss_thumbnails/hypothalamusbydr-260124142231-2b48143d-thumbnail.jpg?width=640&height=640&fit=bounds)
















