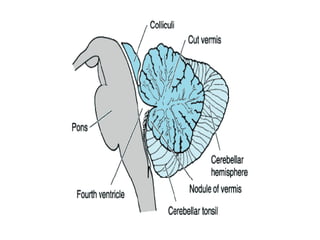
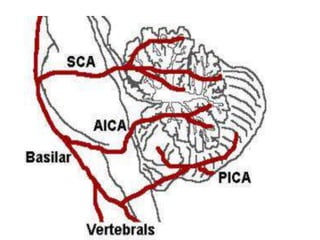
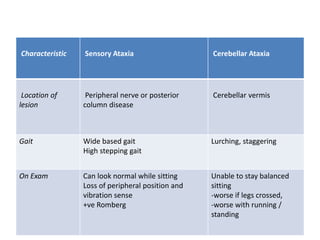
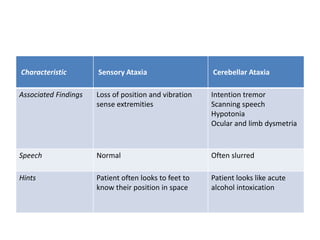
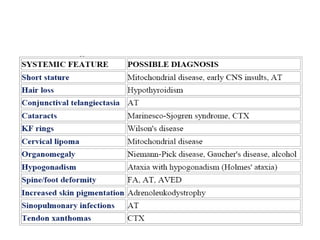
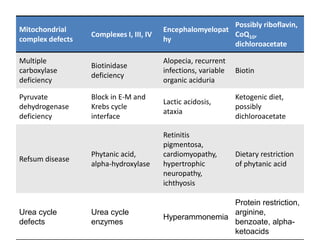
This document discusses ataxia in children. It describes the different types of ataxia including sensory ataxia and cerebellar ataxia. It outlines the characteristic features, locations of lesions, physical exam findings, and hints to differentiate between sensory and cerebellar ataxia. The document also provides guidance on evaluating a child with ataxia including taking a thorough history, performing a full physical and neurological exam, ordering appropriate tests and imaging, and considering possible consultations. Common causes of ataxia in childhood are discussed such as congenital, degenerative/genetic, infectious, metabolic, neoplastic, toxic, traumatic and vascular etiologies.

























![Hereditary Ataxias
• Group1-
caused by enzymatic
defects,
autosomal recessive
manner and
typically present in
childhood; fortunately,
many of these are now
treatable
• Group 2-
progressive degenerative
ataxias
AR-Friedreich ataxia
autosomal dominant
spinocerebellar ataxias,
episodic
ataxias,dentatorubropallidol
uysian atrophy [DRPLA])
the very rare X-linked
ataxias.](https://image.slidesharecdn.com/ataxia-200605145854/85/Ataxia-32-320.jpg)













![• Laboratory Abnormalities in A-T
• Elevated and slowly increasing serum alpha-fetoprotein
levels after two years of age
• Low serum levels of immunoglobulins (IgA, IgG, IgG
subclasses, IgE) and lymphopenia (particularly affecting
T-lymphocytes)
• Spontaneous and X-ray induced chromosomal breaks
and rearrangements in cultured lymphocytes and
fibroblasts
• Reduced survival of cultured lymphocytes and
fibroblasts after exposure to ionizing radiation [209]
• Cerebellar atrophy detected by MRI](https://image.slidesharecdn.com/ataxia-200605145854/85/Ataxia-51-320.jpg)






























































































