Download as PDF, PPTX



![Why Wannier Functions?
Advantages of Wannier function basis set:
<Explicit form of the orbitals
forming complete basis set
< Localized orbitals
< Orbitals are centered on atoms
Wannier functions in real space [1]:
[1] G.H. Wannier, Phys. Rev. 52, 192 (1937)
Bloch functions
like in Hubbard model
Uncertainty of WF definition
for a many-band case:
Unitary matrix](https://image.slidesharecdn.com/anisimov-170124101033/75/Density-Functional-and-Dynamical-Mean-Field-Theory-DFT-DMFT-method-and-its-application-to-real-materials-7-2048.jpg)
![Wannier functions and projection
Eigenvector
element
WF in k-space – projection of the set of trial functions [2] (atomic
orbitals) into Bloch functions subspace :
Bloch functions in DFT basis
(LMTO or plane waves):
coefficients of WF expansion in LMTO-orbitals:
Bloch sums of
LMTO orbitals
[2] D.Vanderbildt et al, Phys. Rev.B 56, 12847 (1997)](https://image.slidesharecdn.com/anisimov-170124101033/75/Density-Functional-and-Dynamical-Mean-Field-Theory-DFT-DMFT-method-and-its-application-to-real-materials-8-2048.jpg)
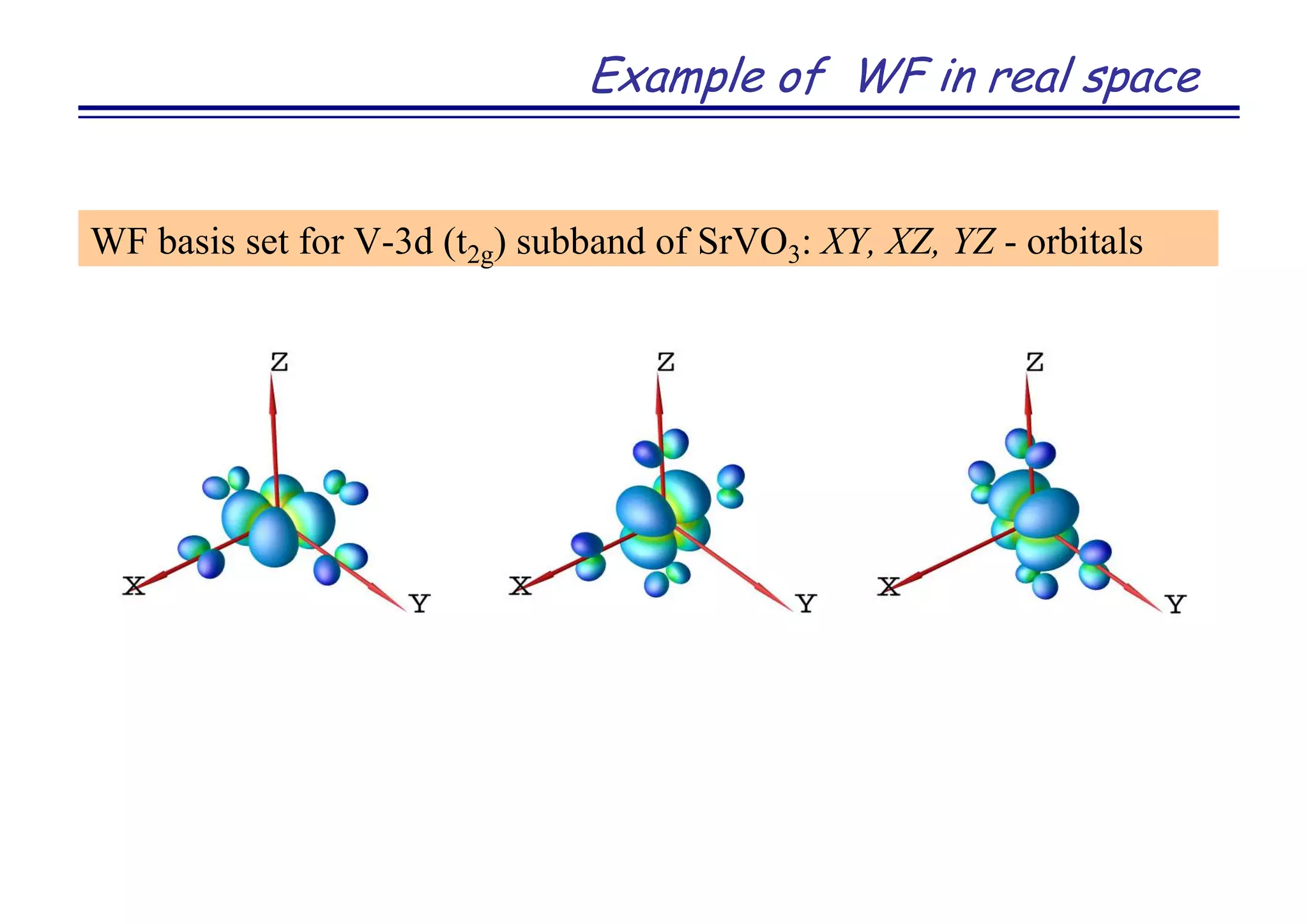
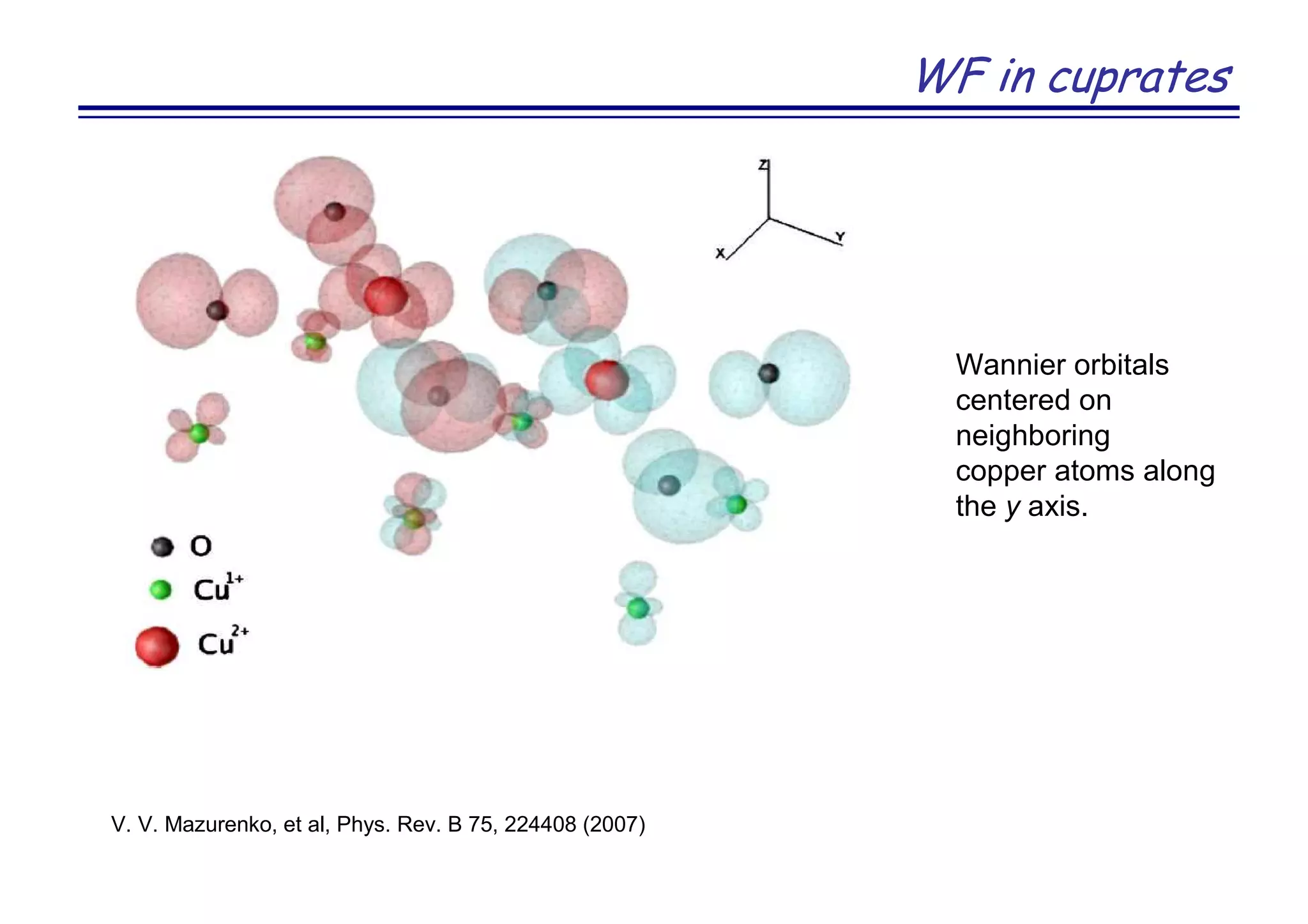
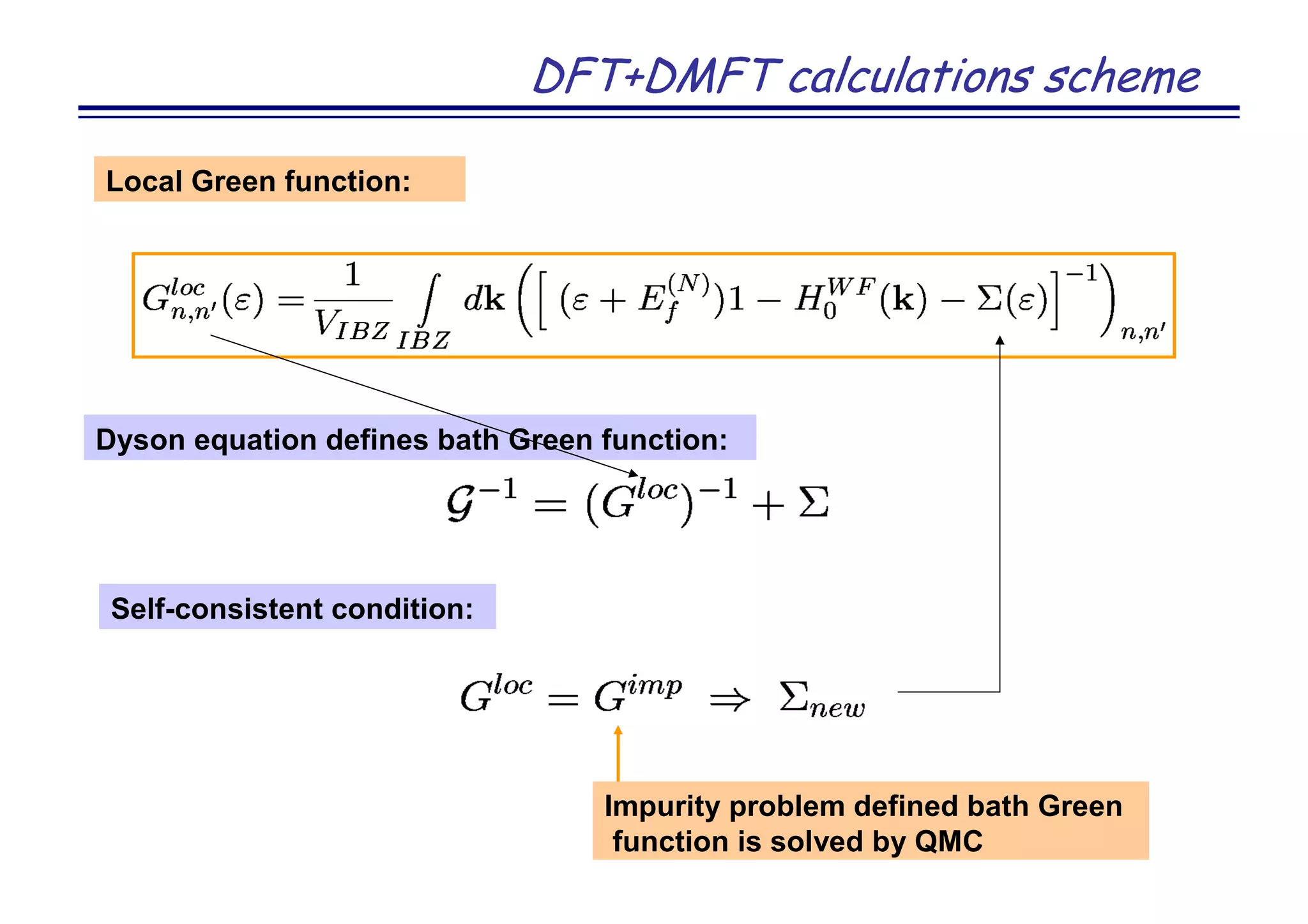
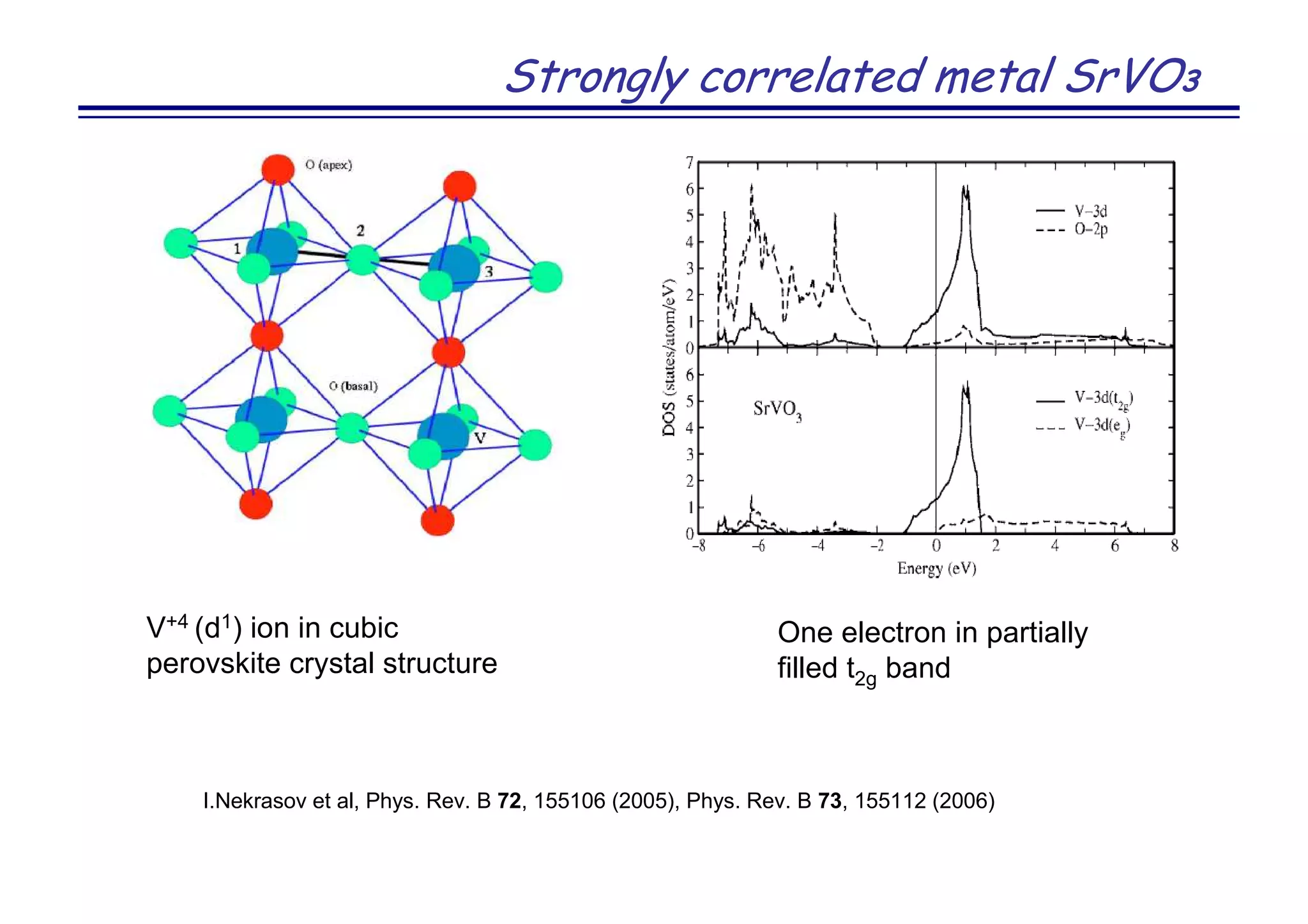
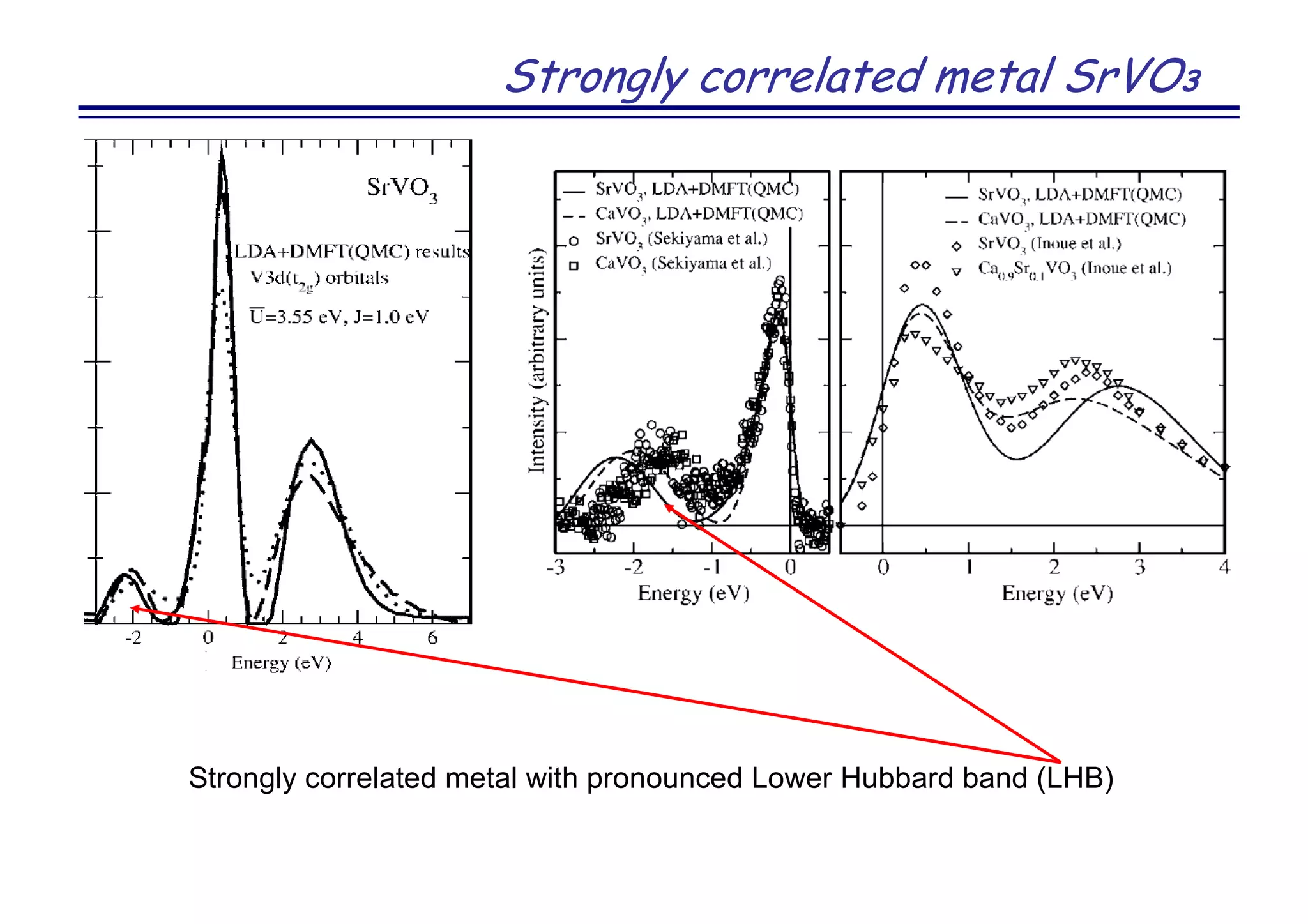
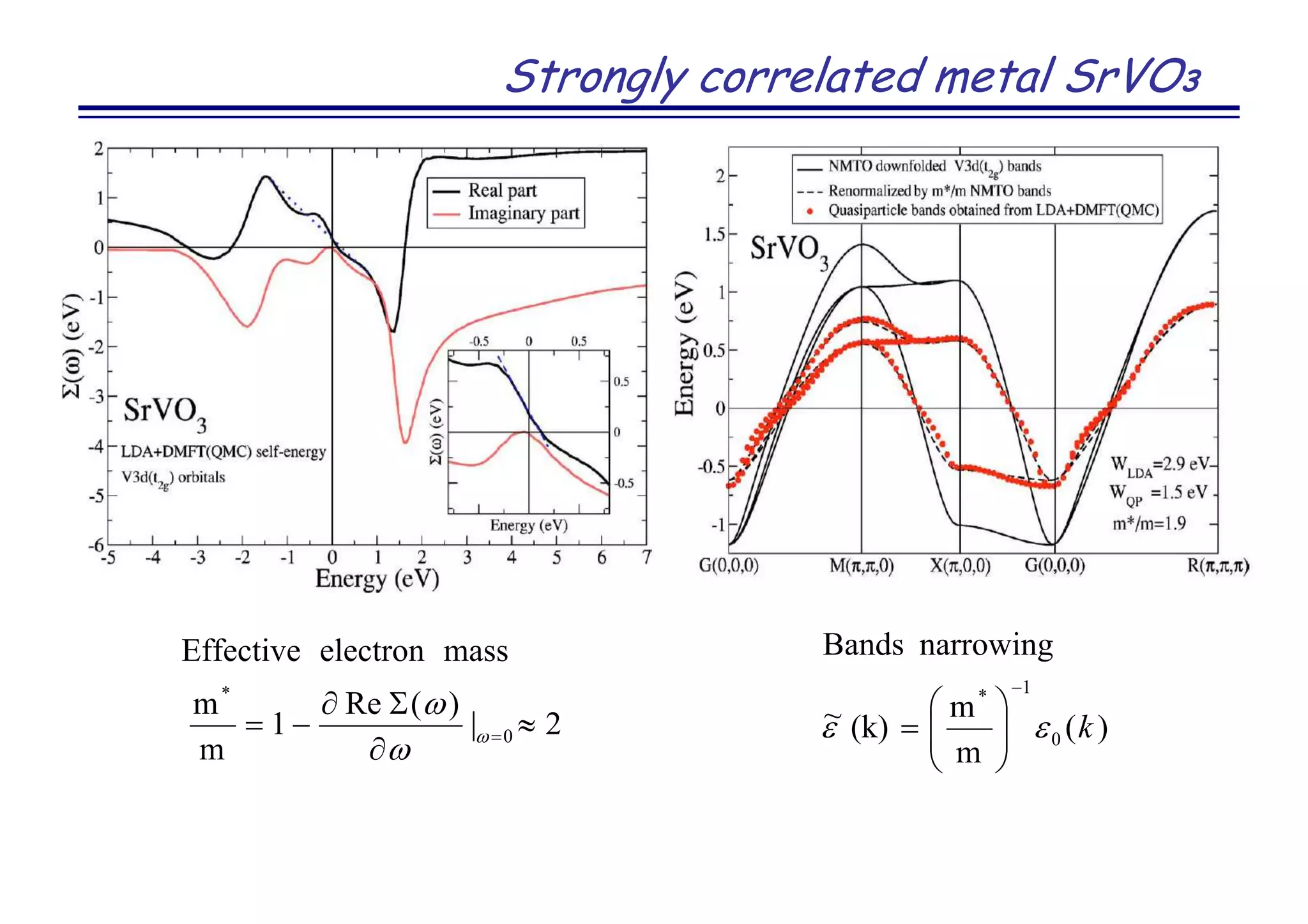
![Strongly correlated metal SrVO3
[ ] 1
0 (k)H-)(-TrIm
1
),(AfunctionSpectral
−
Σ−= ωω
π
ωk](https://image.slidesharecdn.com/anisimov-170124101033/75/Density-Functional-and-Dynamical-Mean-Field-Theory-DFT-DMFT-method-and-its-application-to-real-materials-14-2048.jpg)
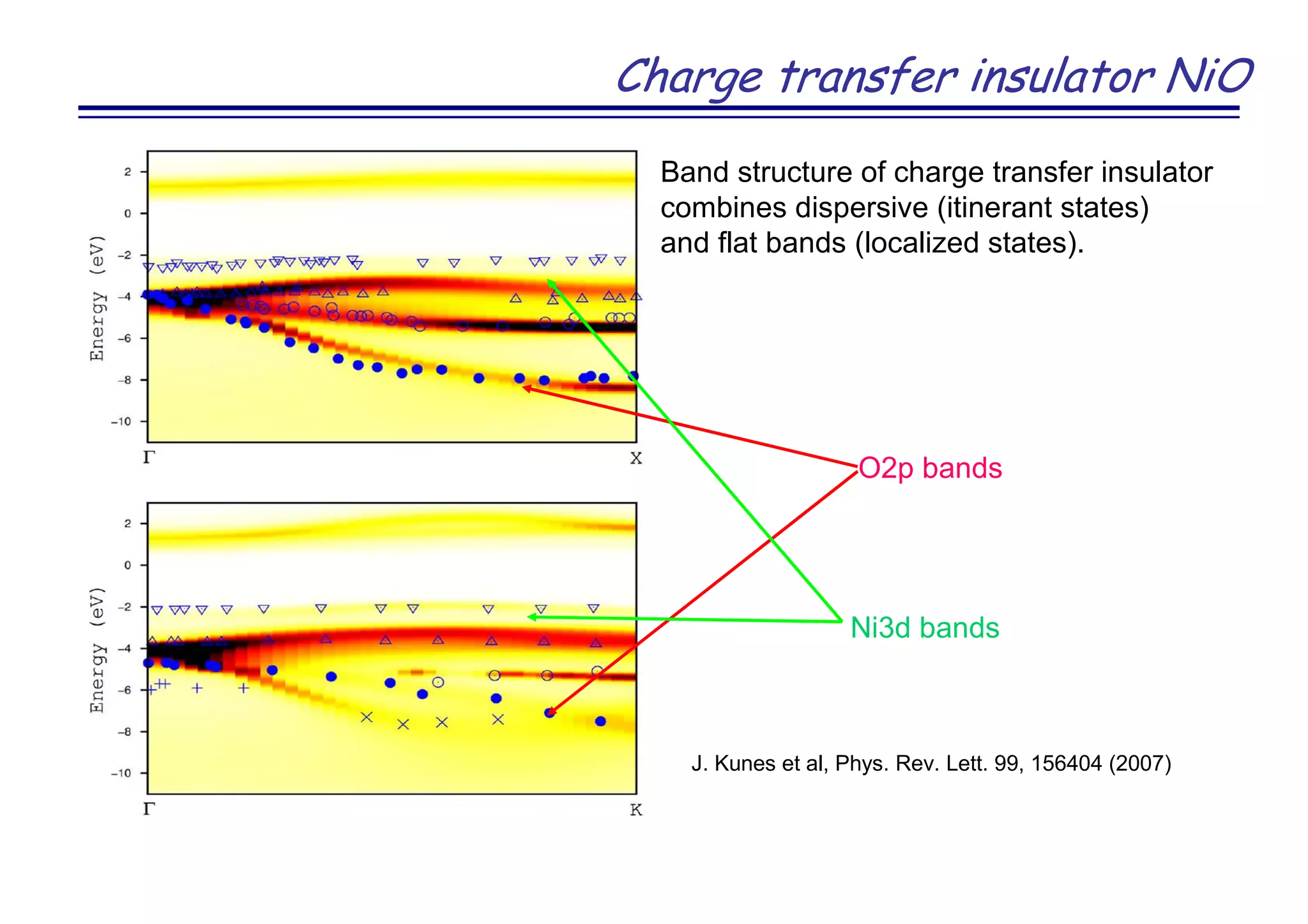
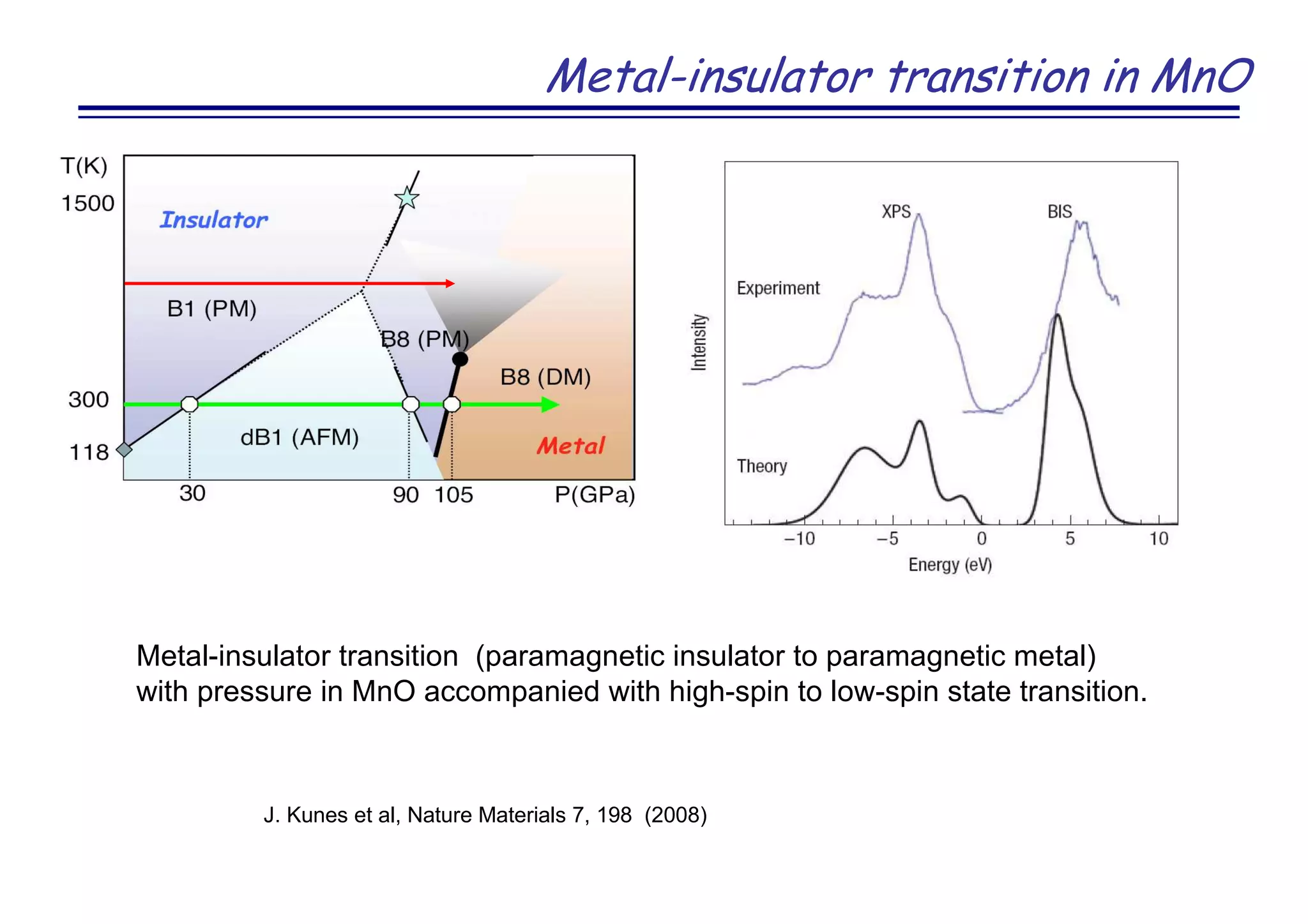
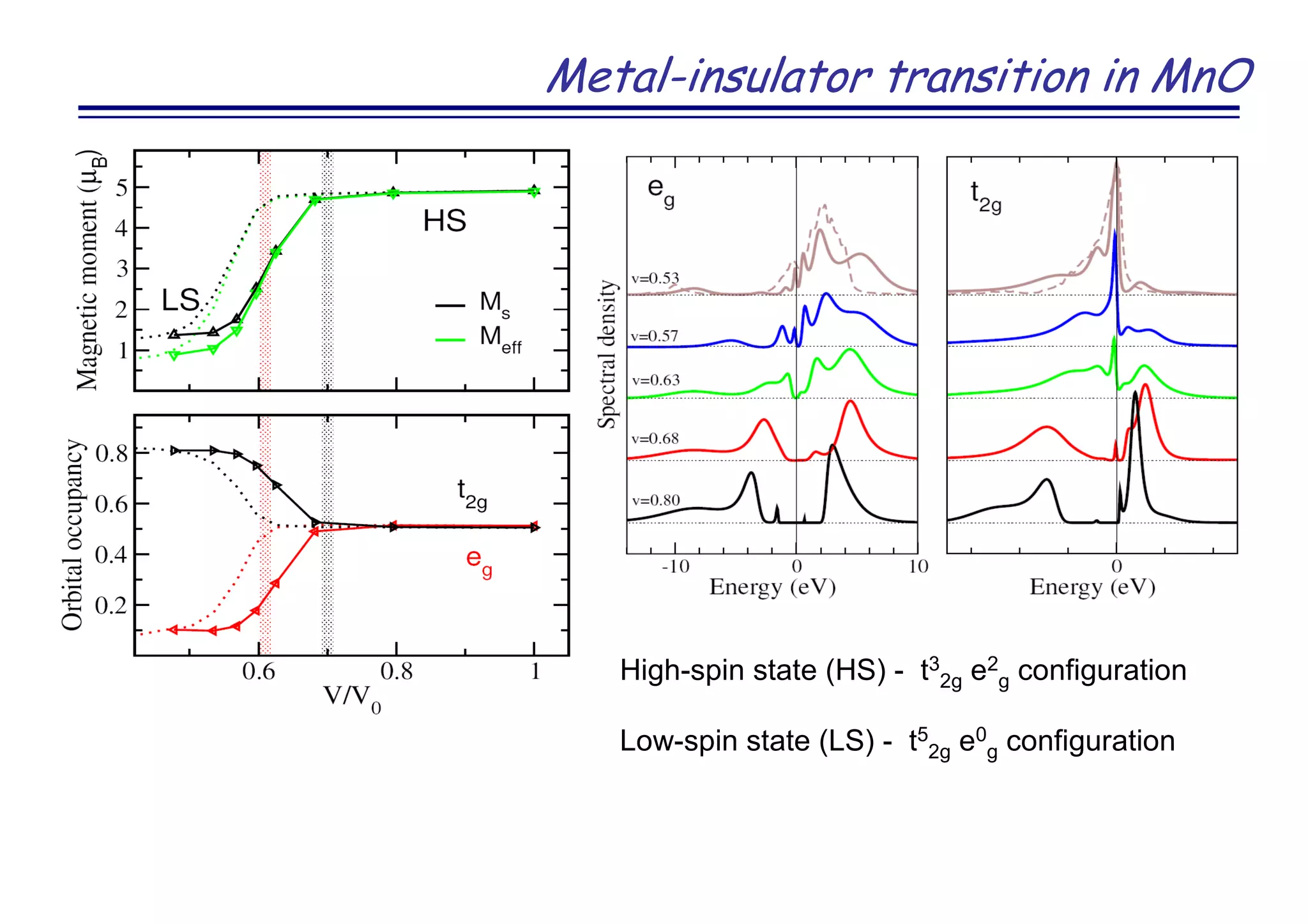

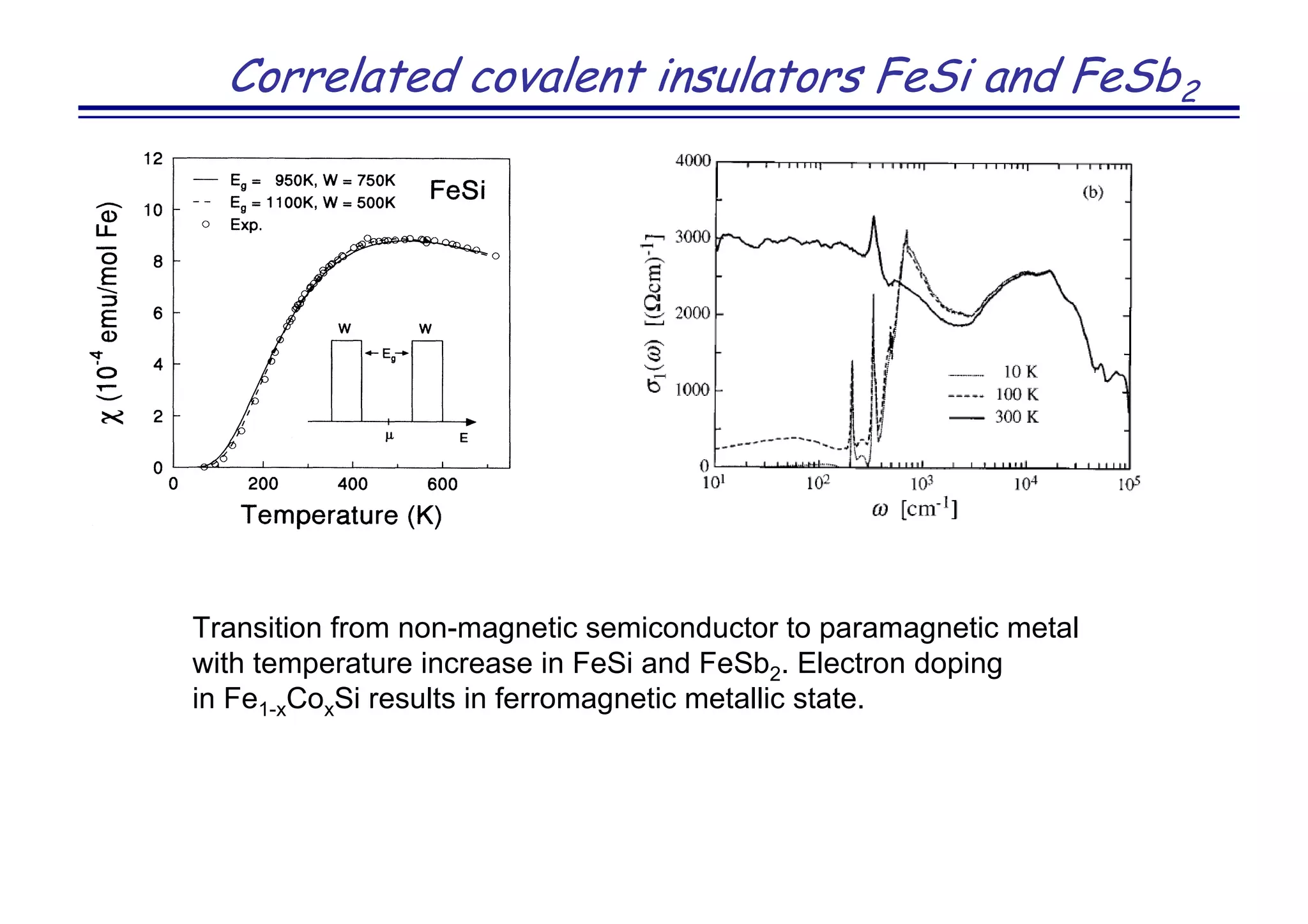
![Novel superconductor LaOFeAs
Tc=26K for F content ~11%
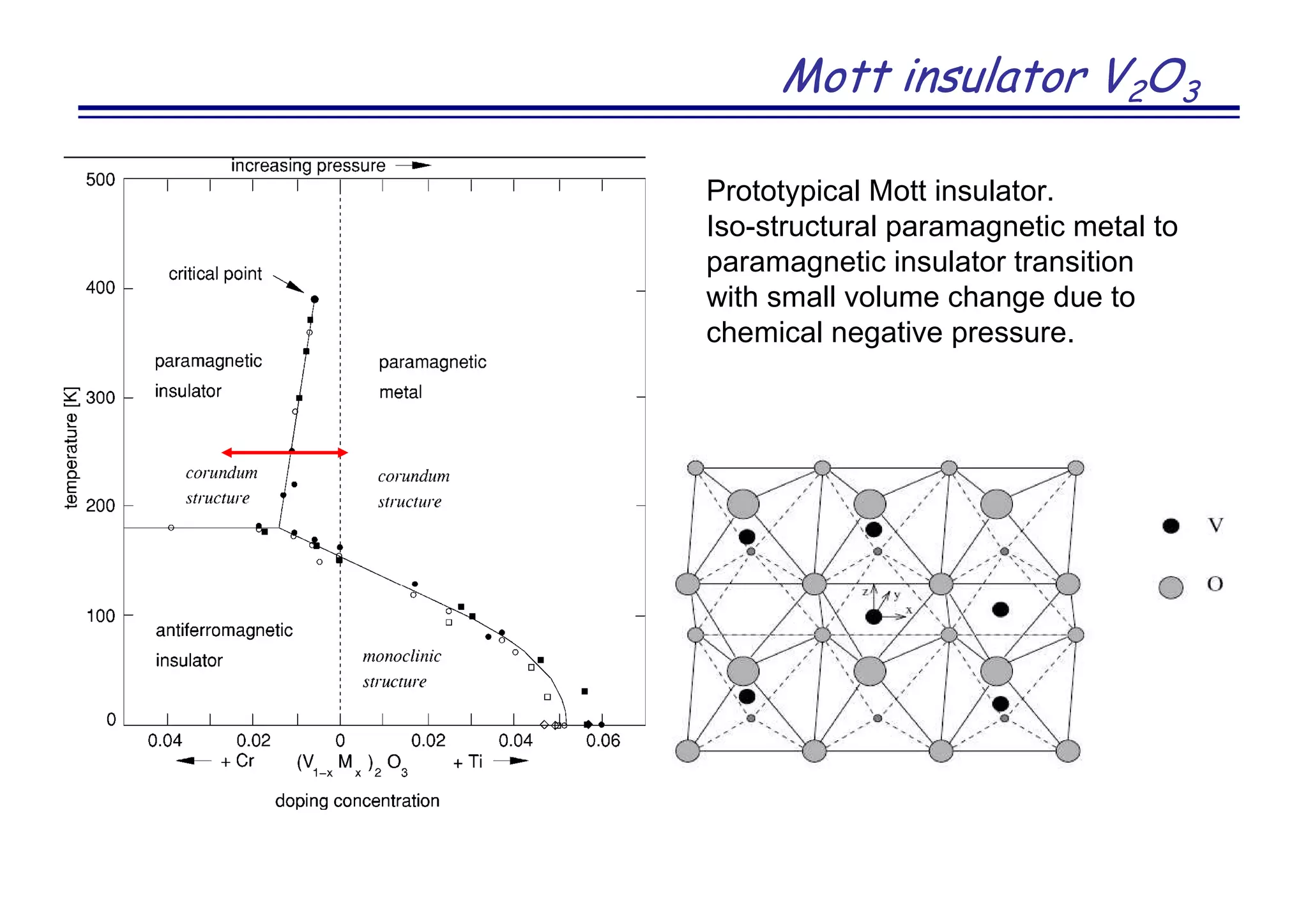
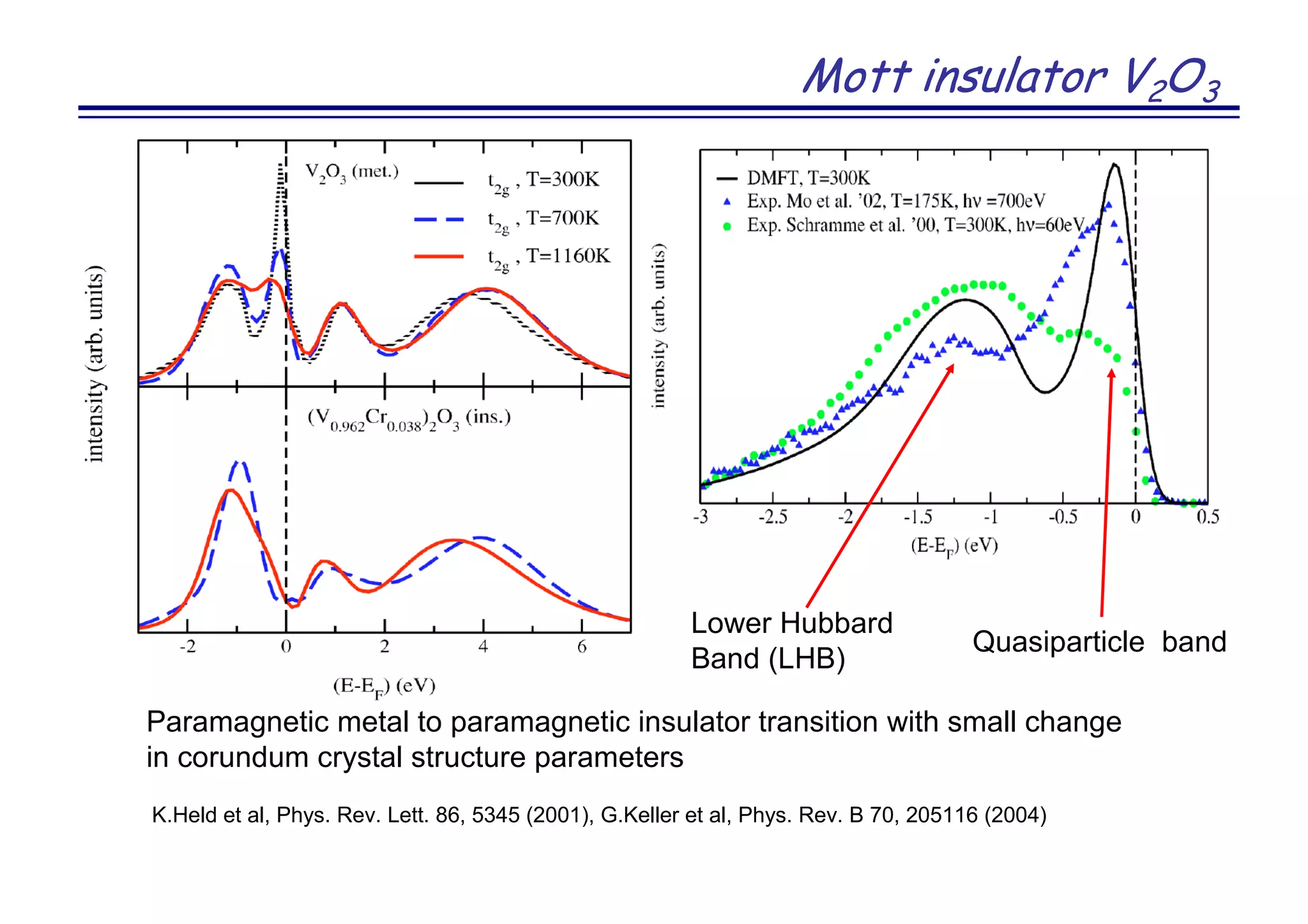
Y. Kanamura et al. J. Am. Chem. Soc. 130, 3296 (2008)]](https://image.slidesharecdn.com/anisimov-170124101033/75/Density-Functional-and-Dynamical-Mean-Field-Theory-DFT-DMFT-method-and-its-application-to-real-materials-28-2048.jpg)
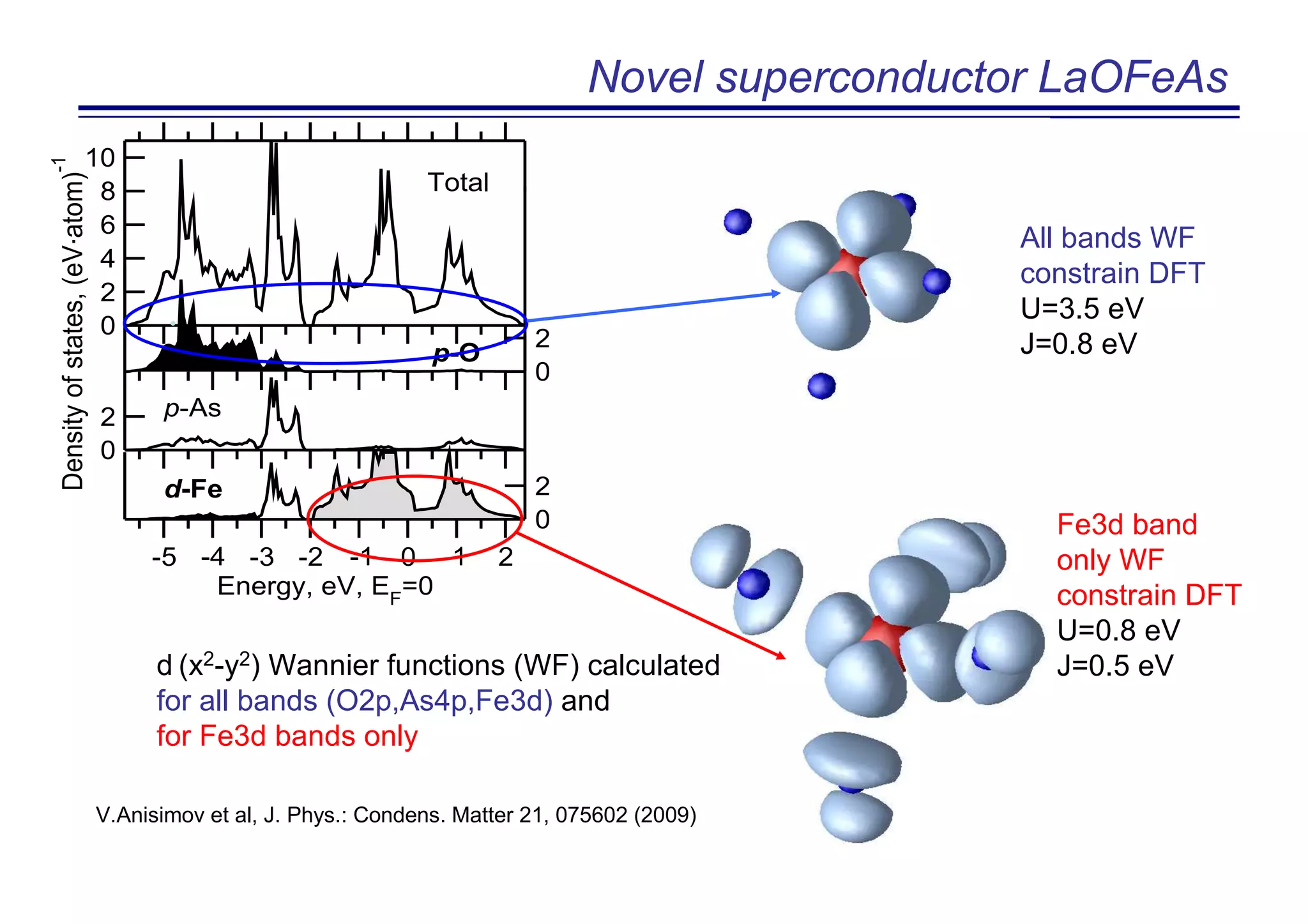
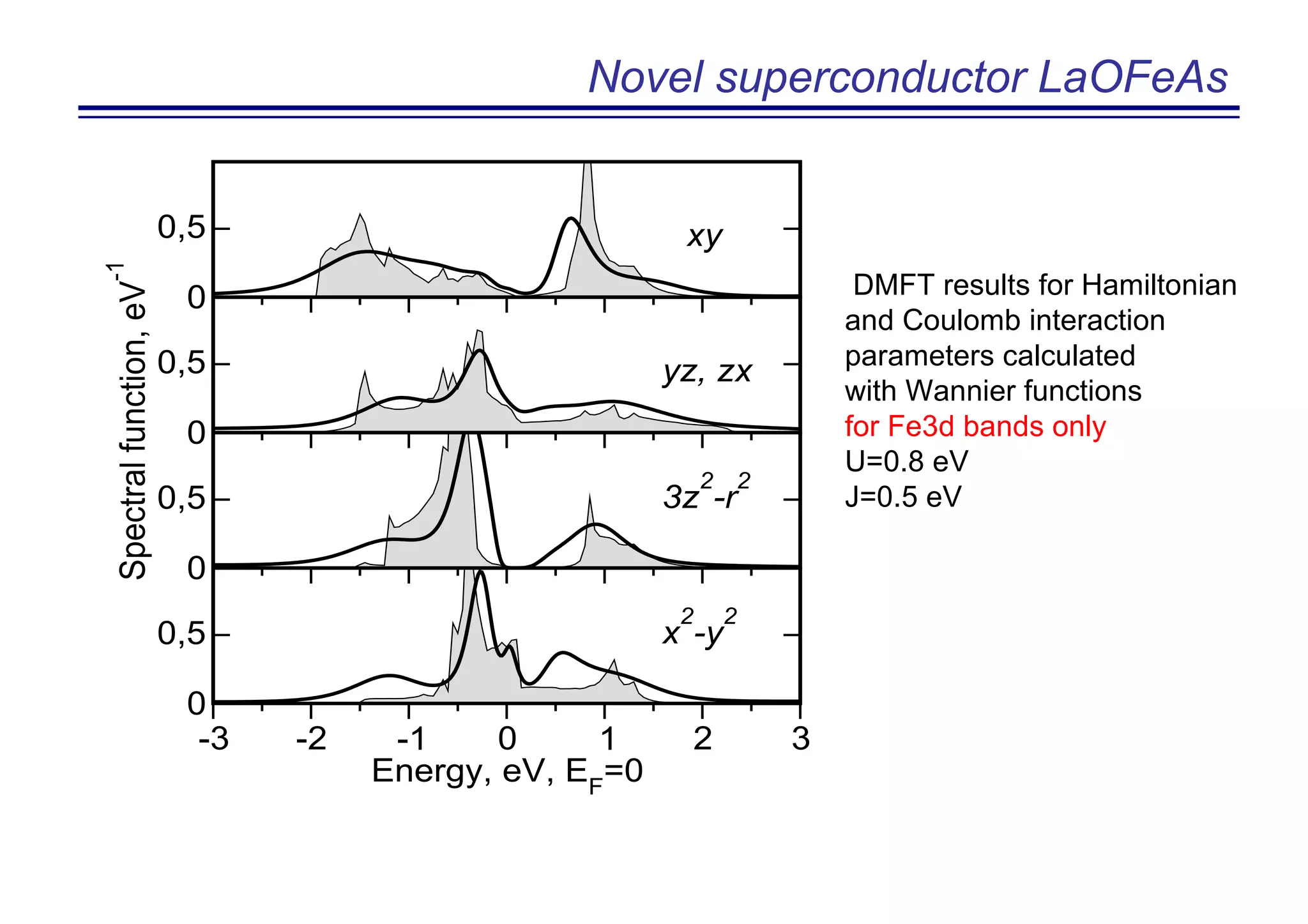
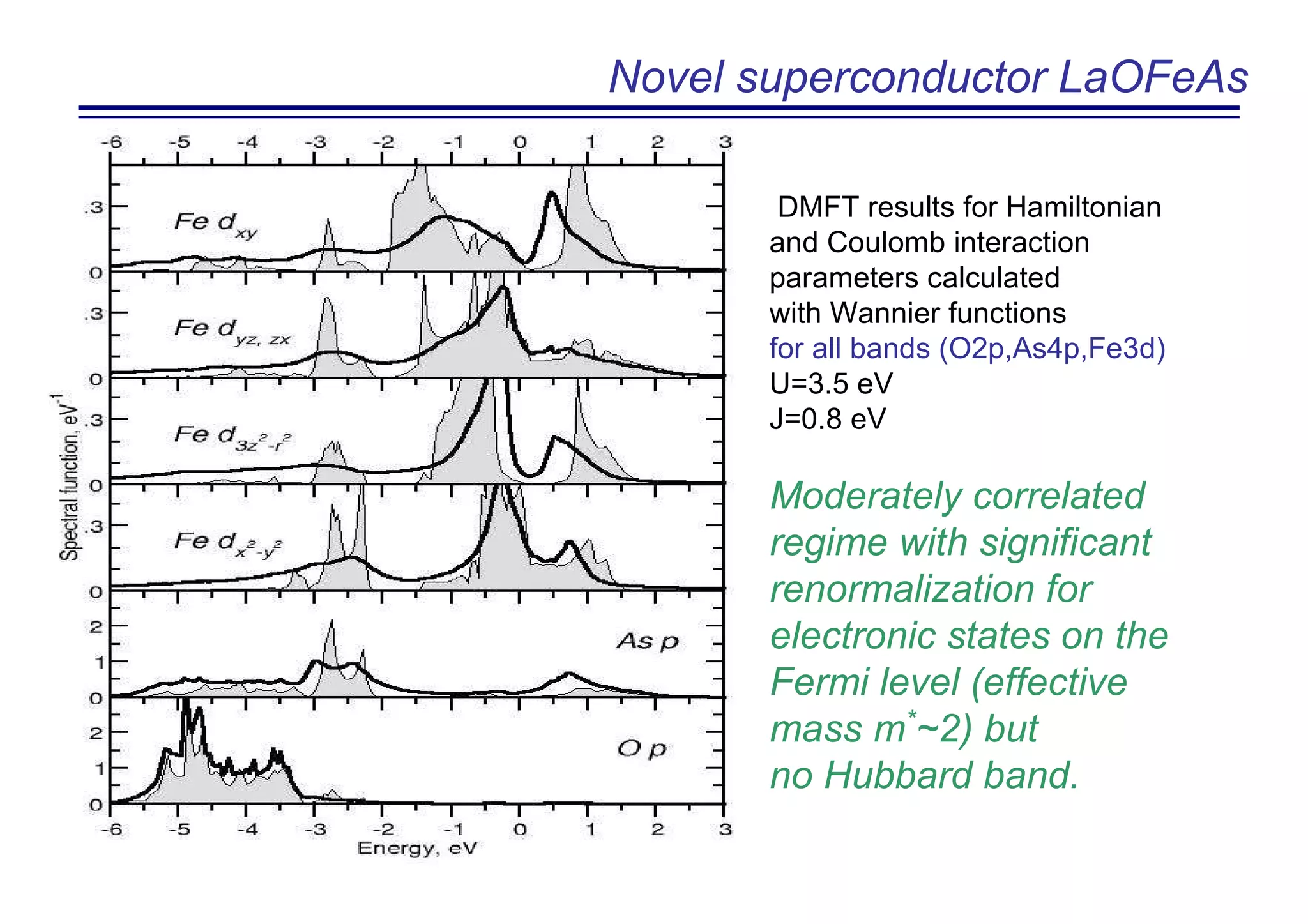
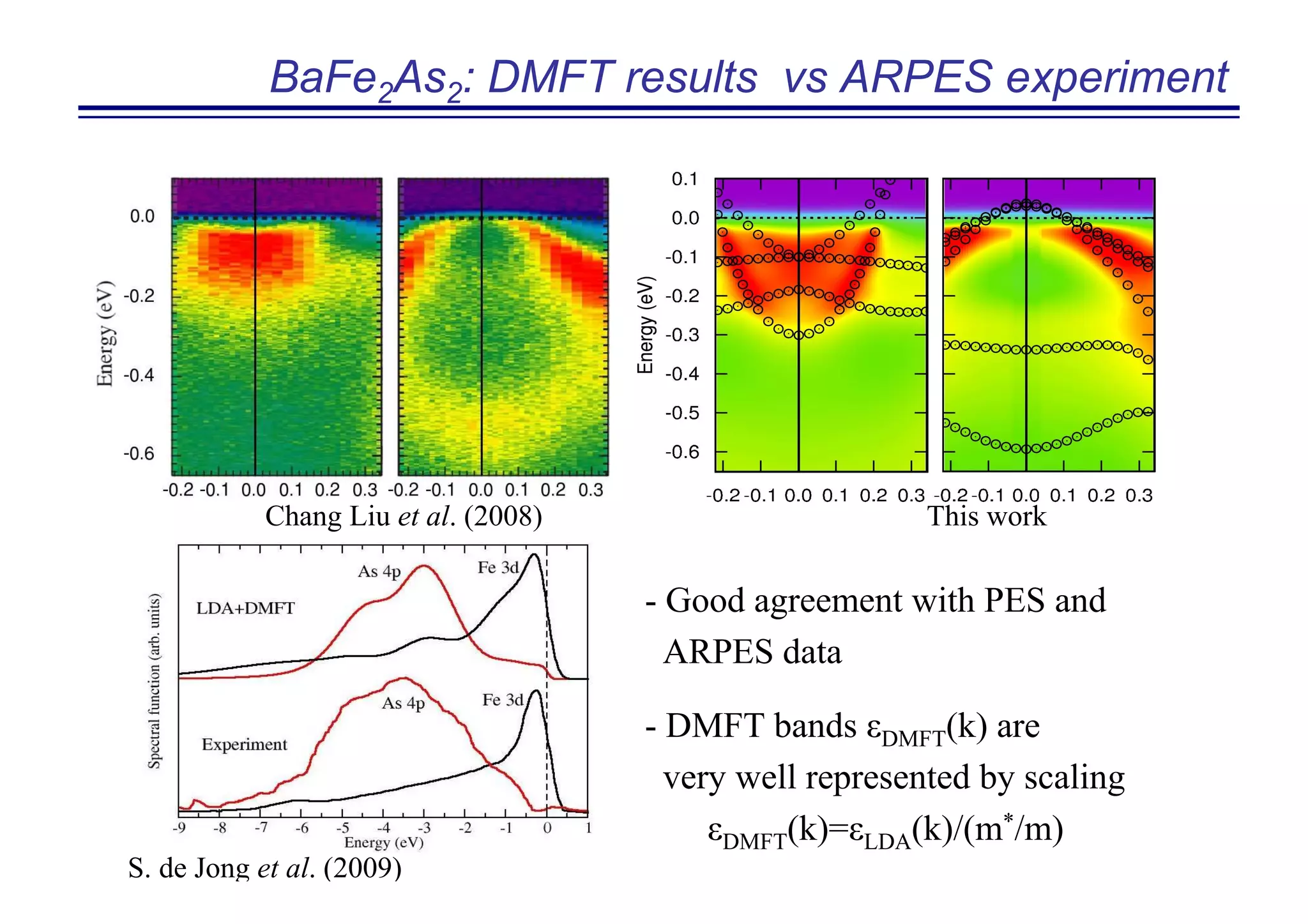
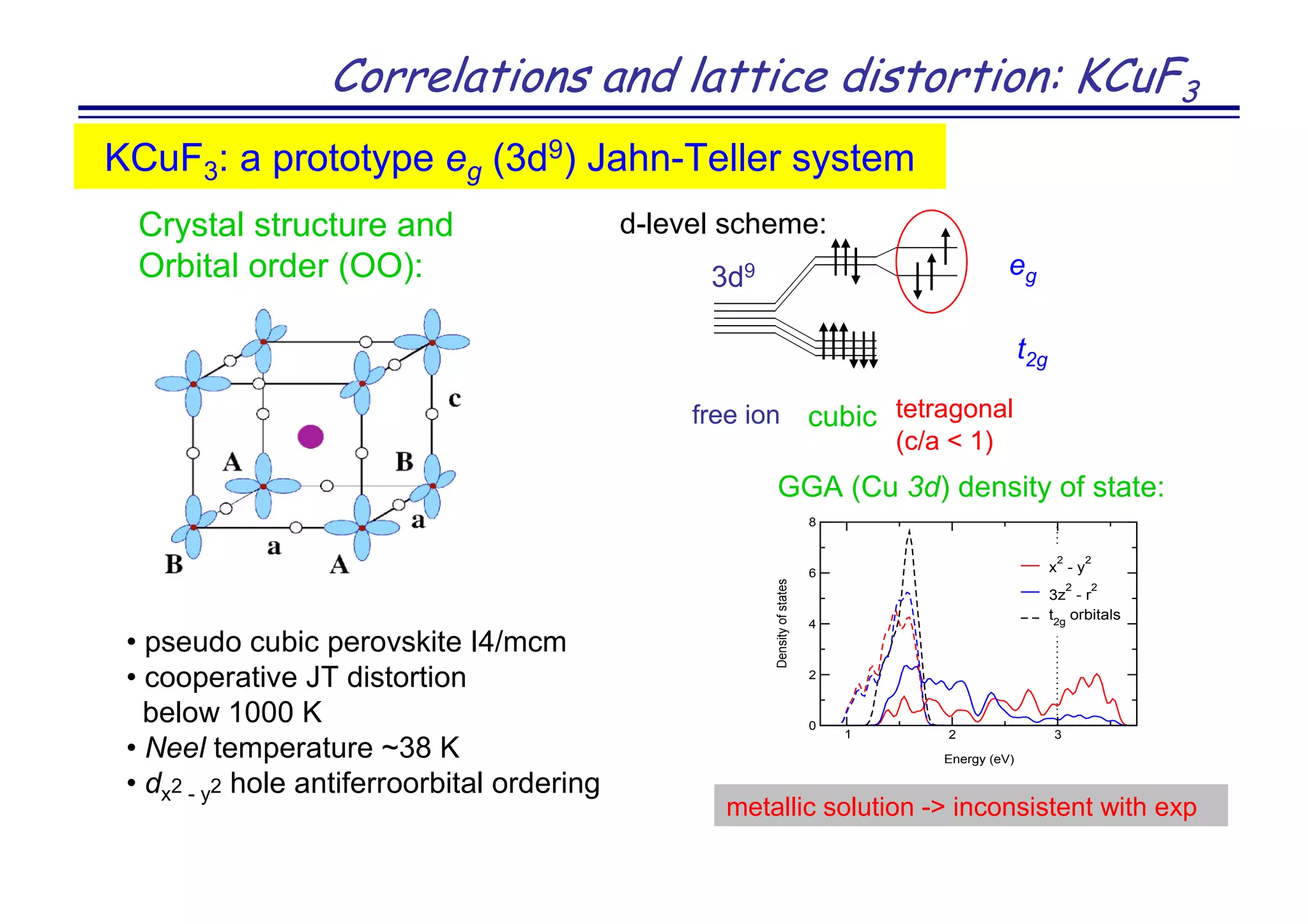
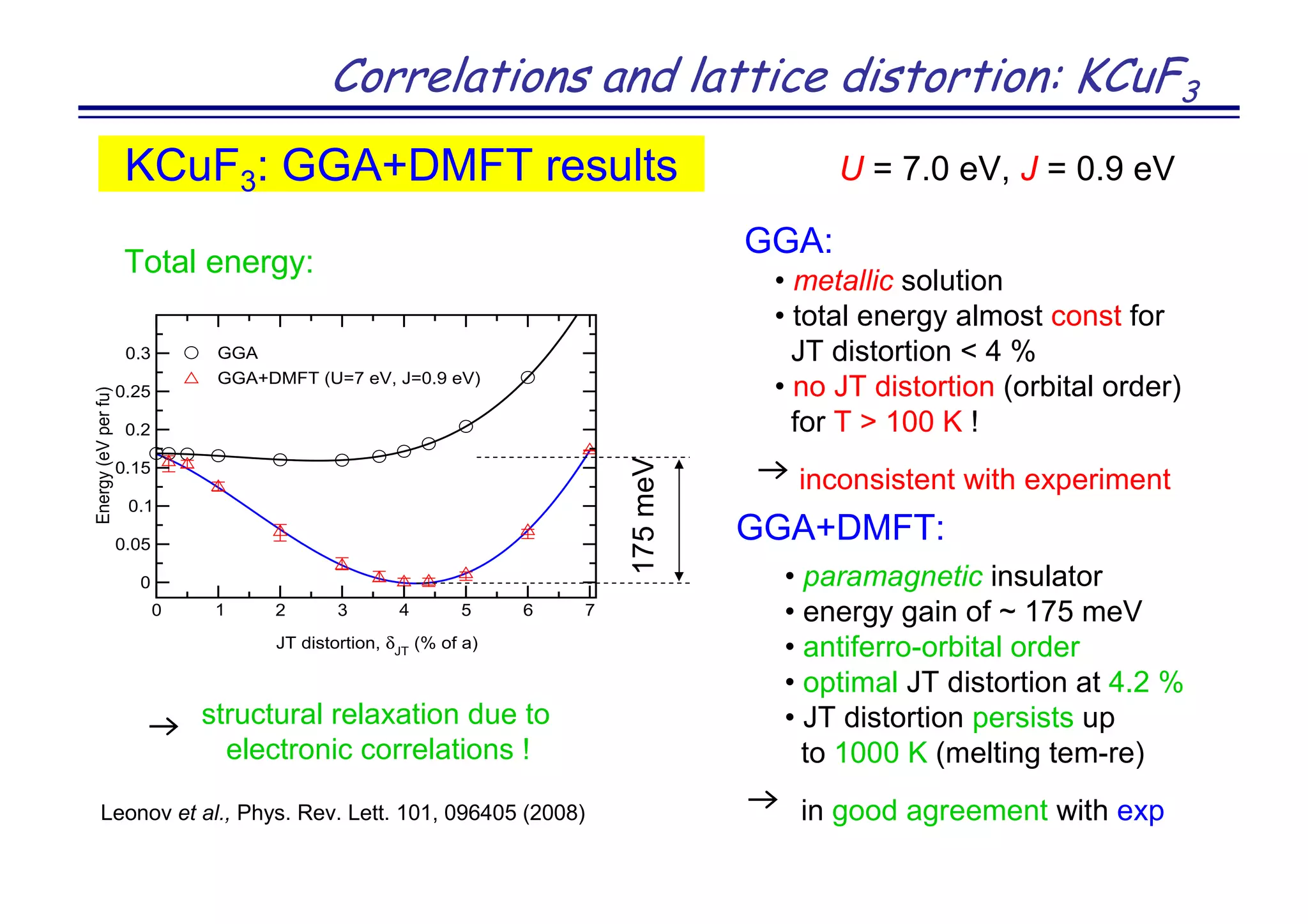
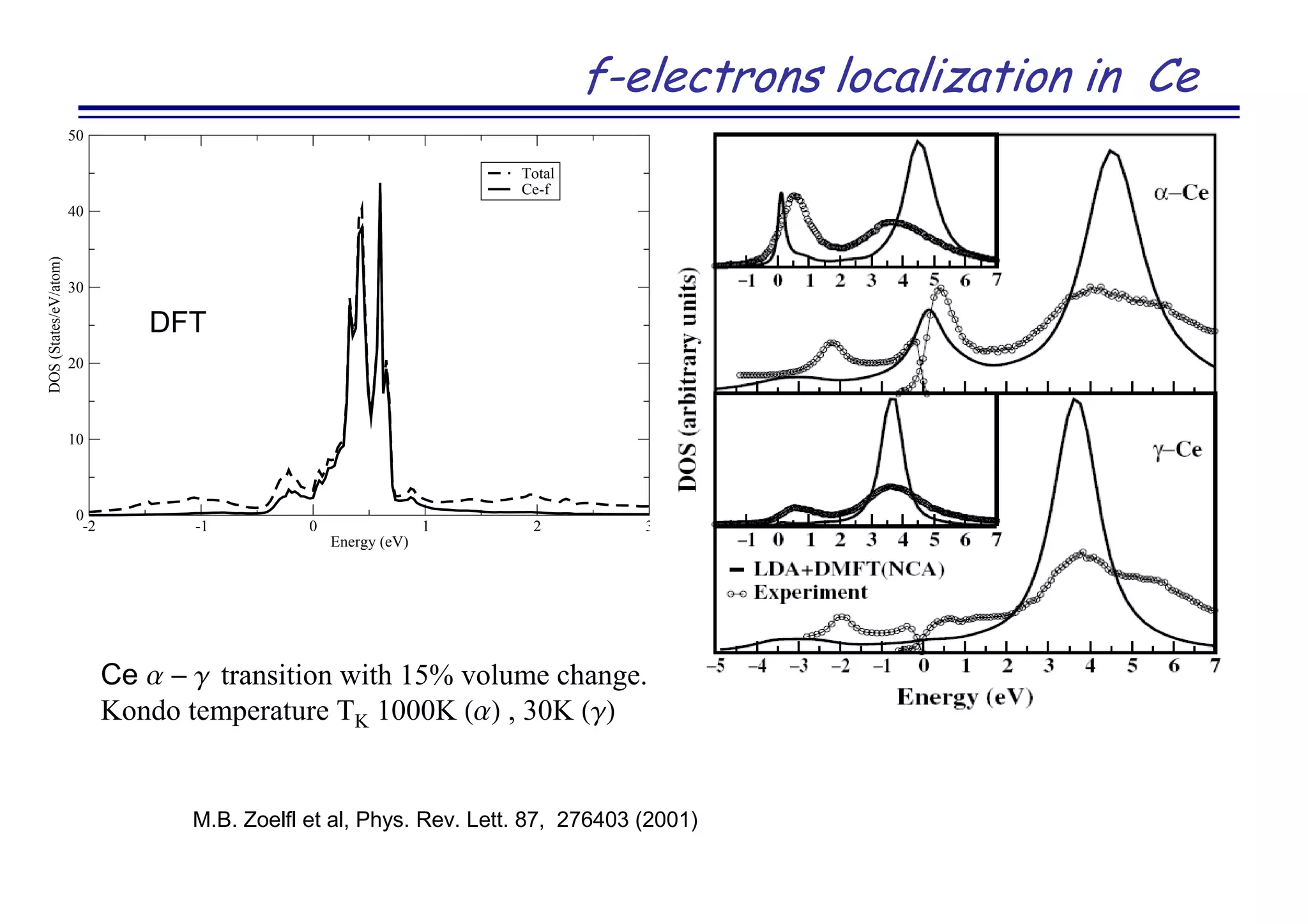


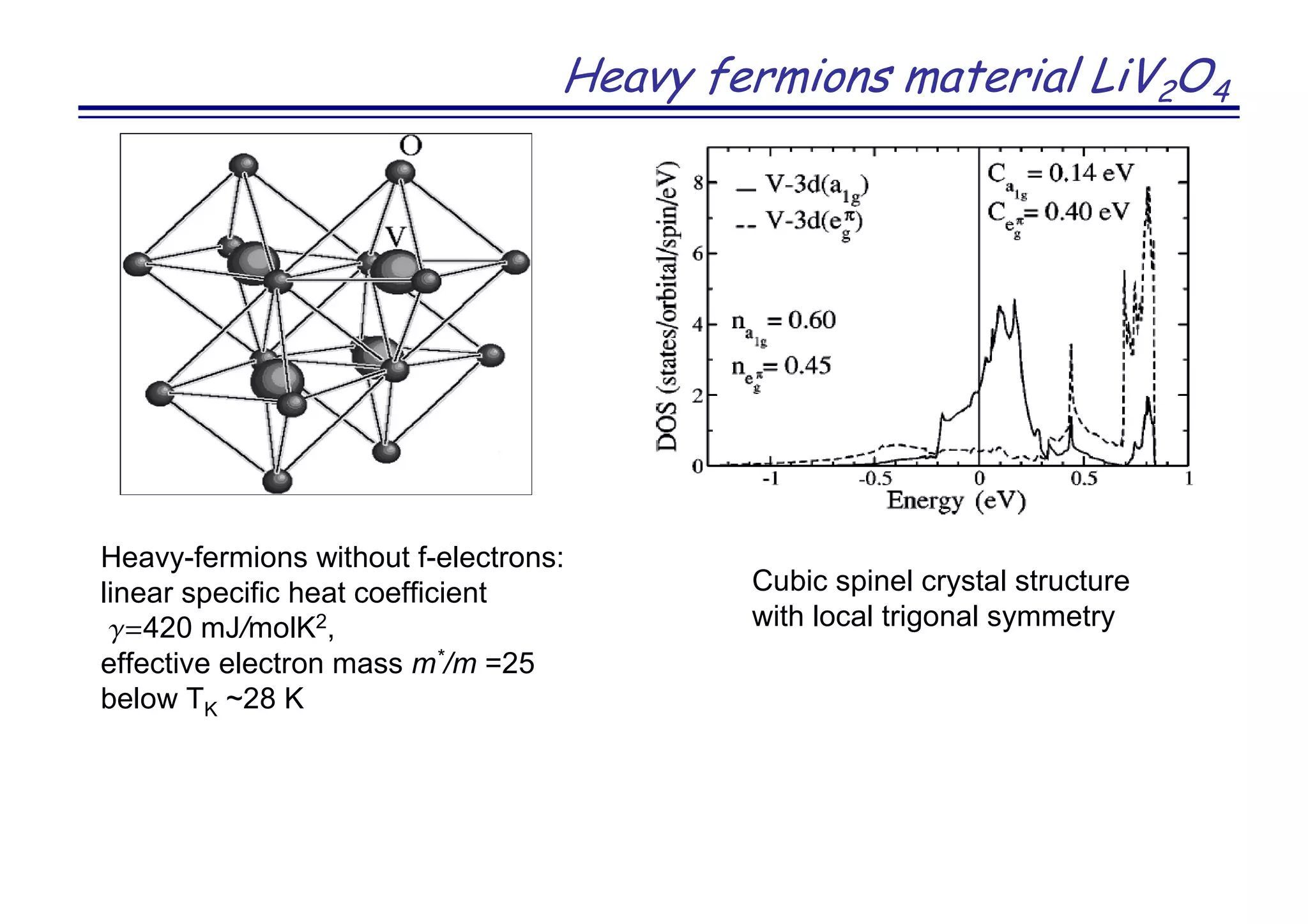
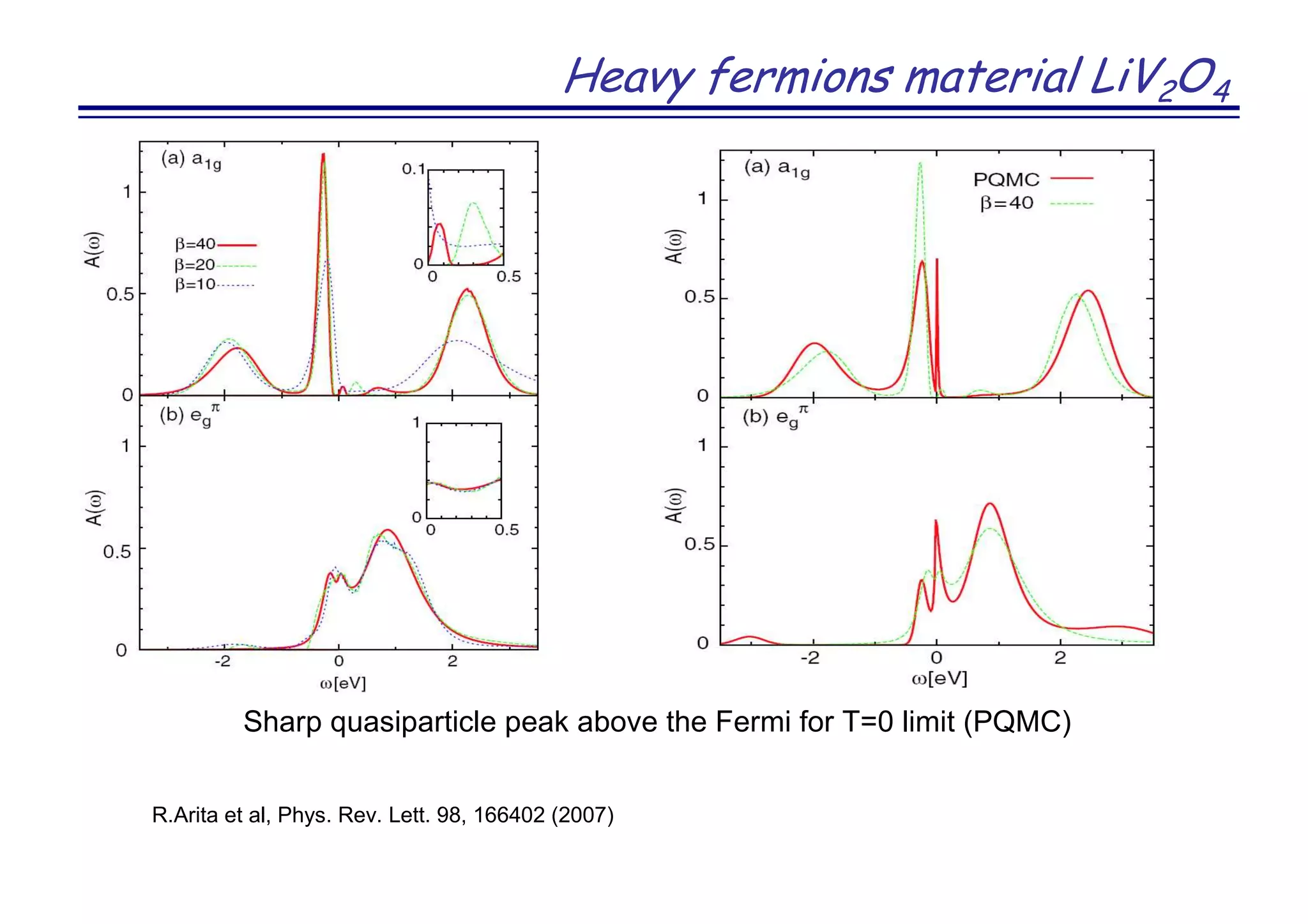
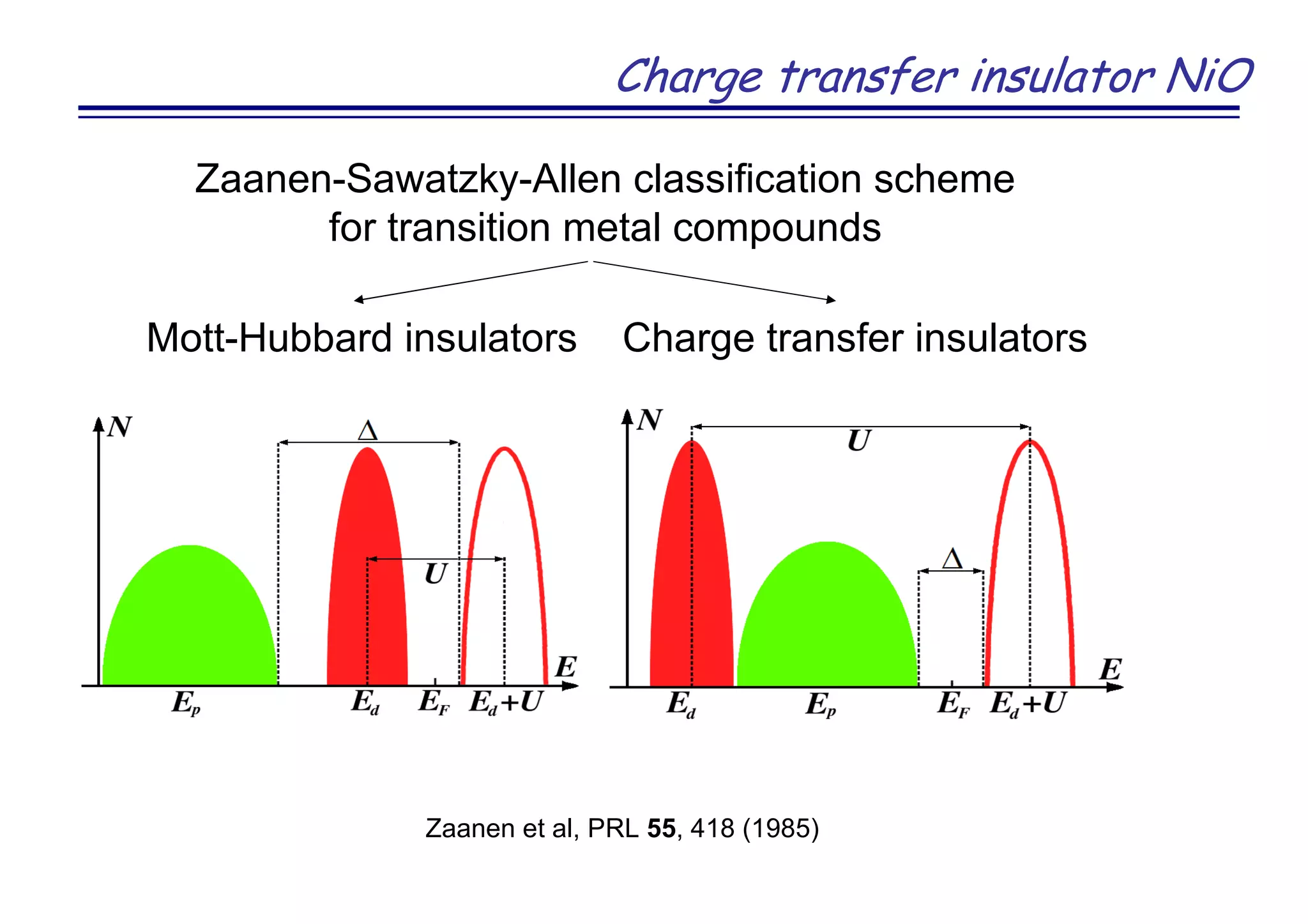
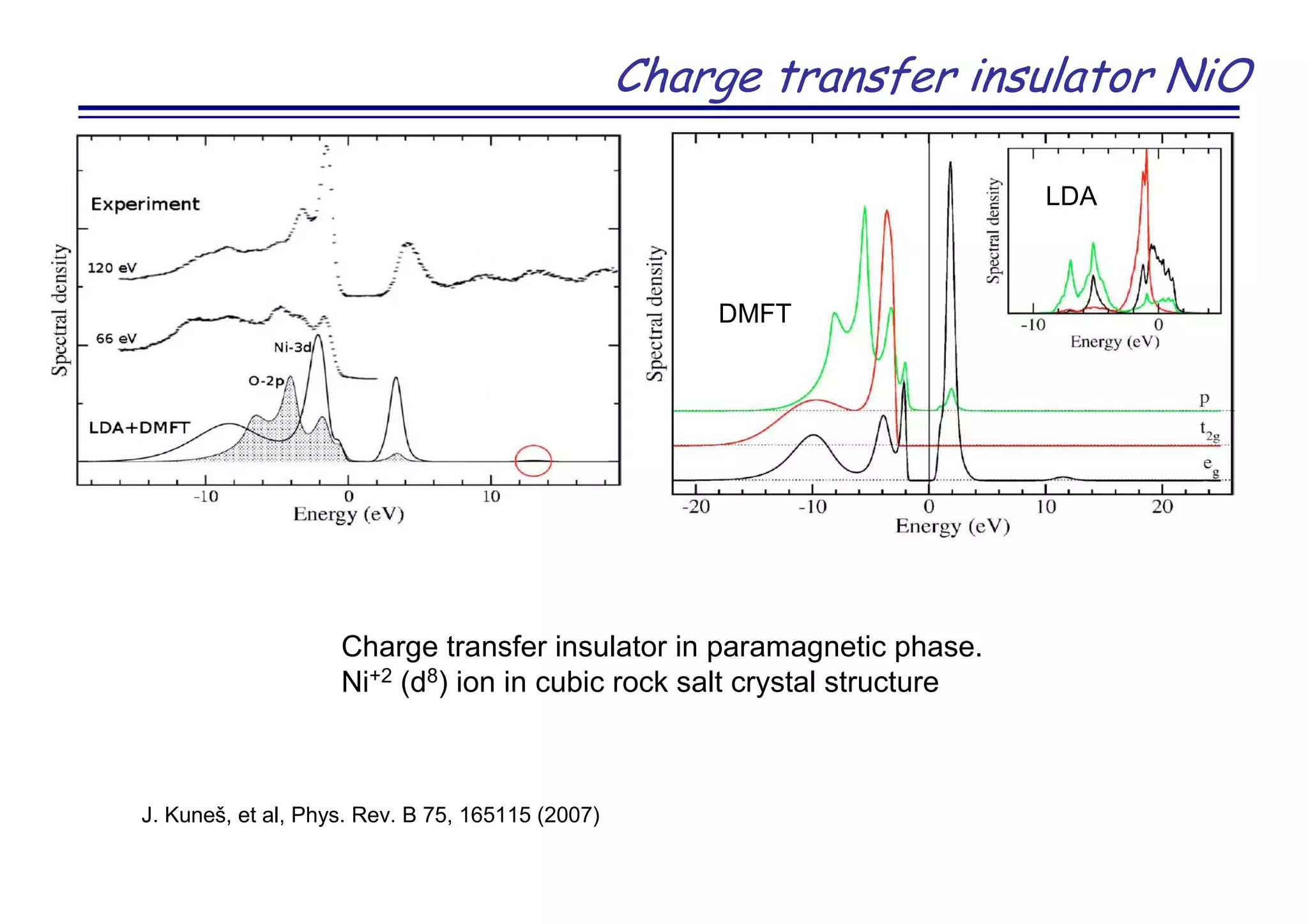
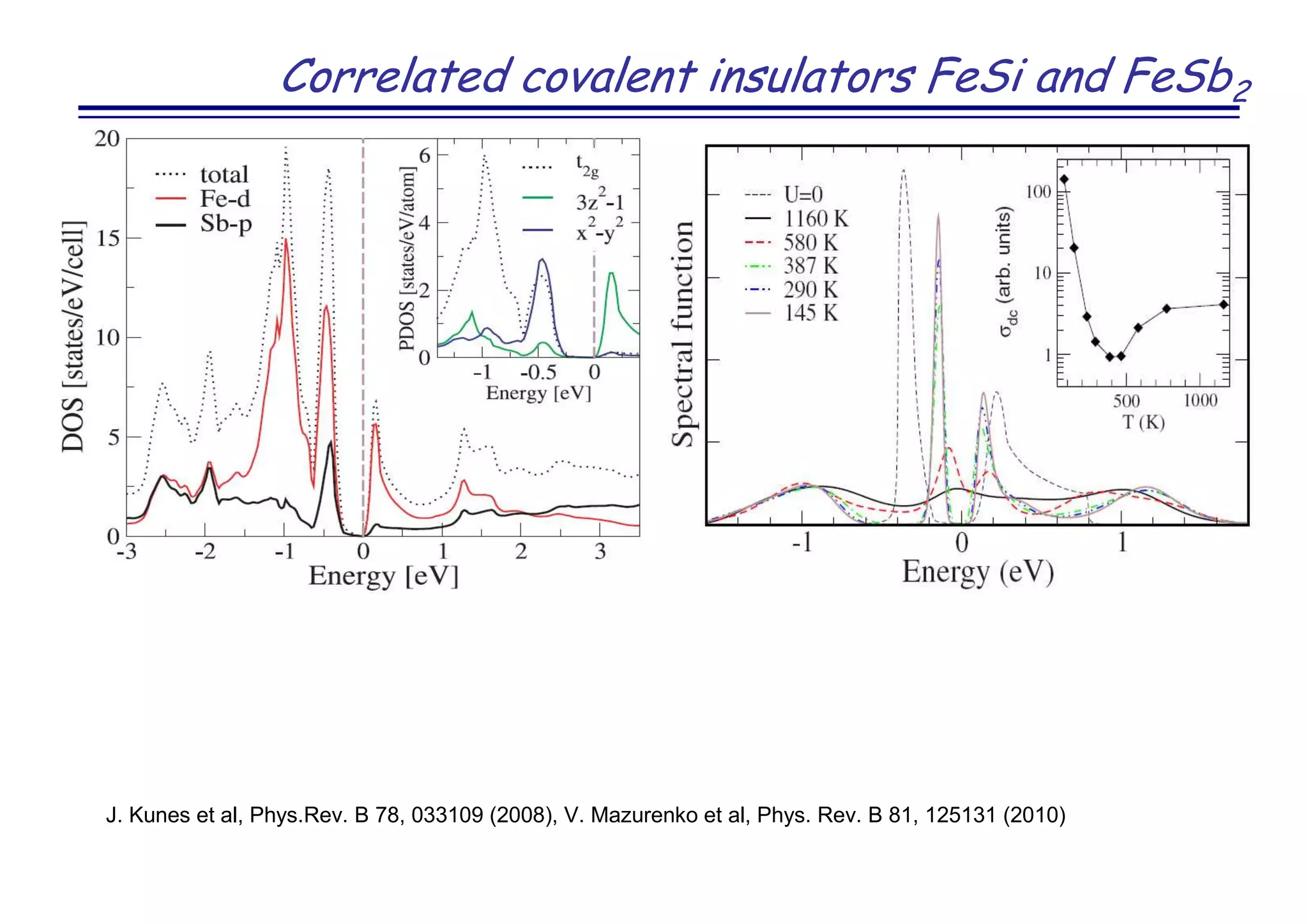
The document discusses the density functional and dynamical mean-field theory (DFT+DMFT) method, focusing on its formulation, application to real materials, and analysis of strongly correlated systems. It highlights the method's effectiveness in predicting properties of various materials, including Mott insulators, charge transfer insulators, and novel superconductors. Key results and examples include metal-insulator transitions and the role of interaction parameters defined through the Wannier functions formalism, showcasing DFT+DMFT as a robust tool for studying correlated electron systems.