Downloaded 15 times
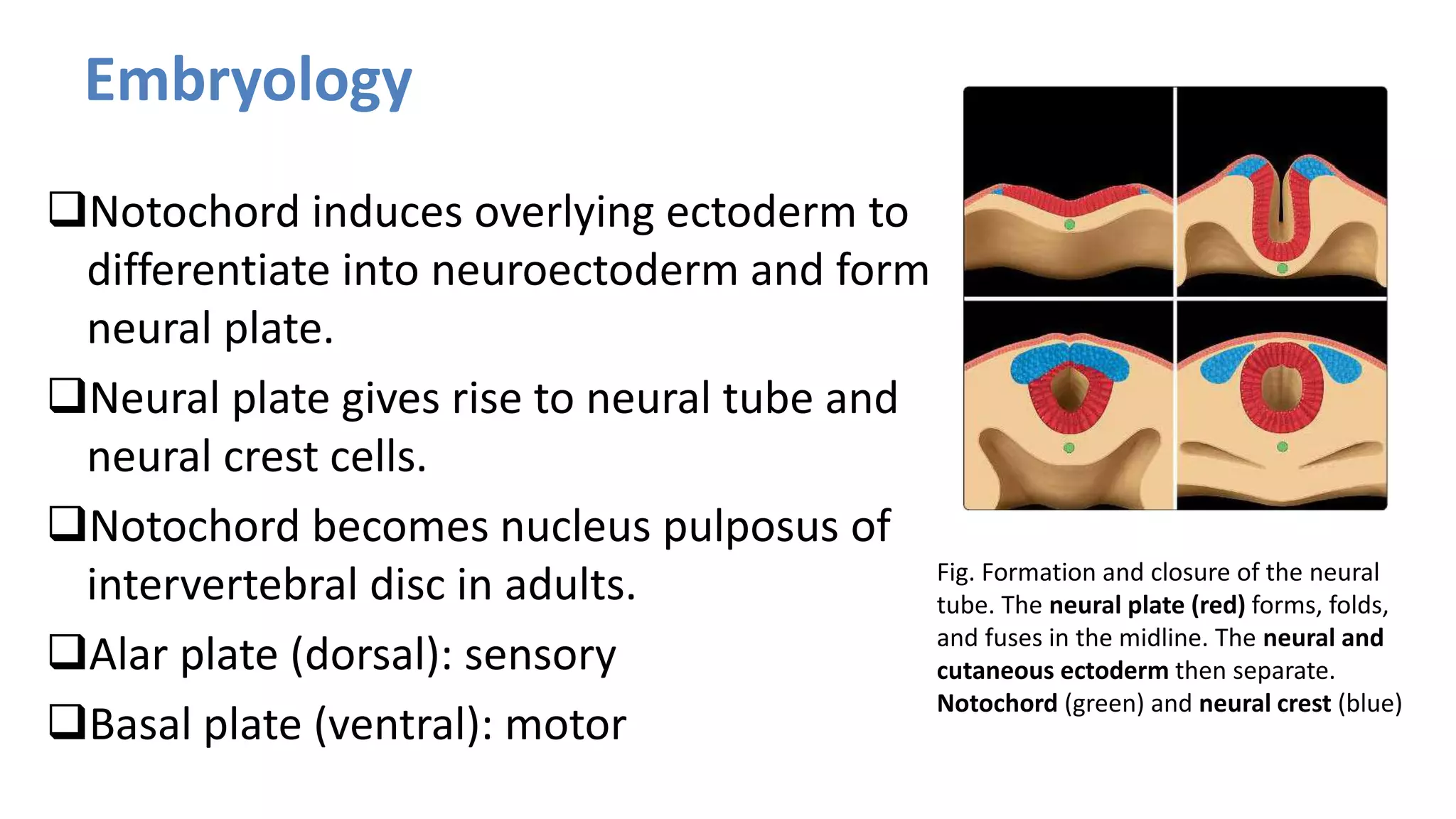






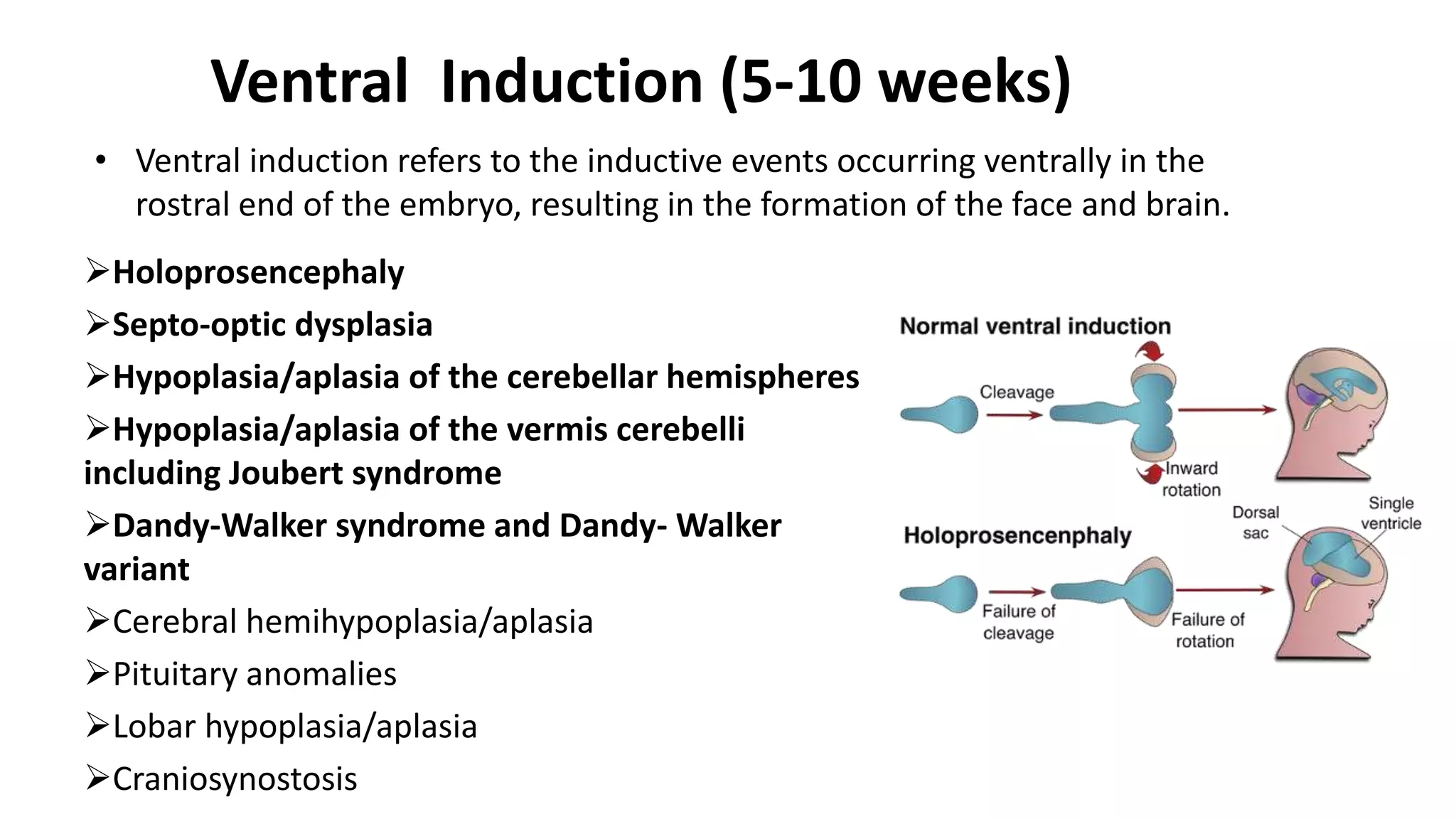





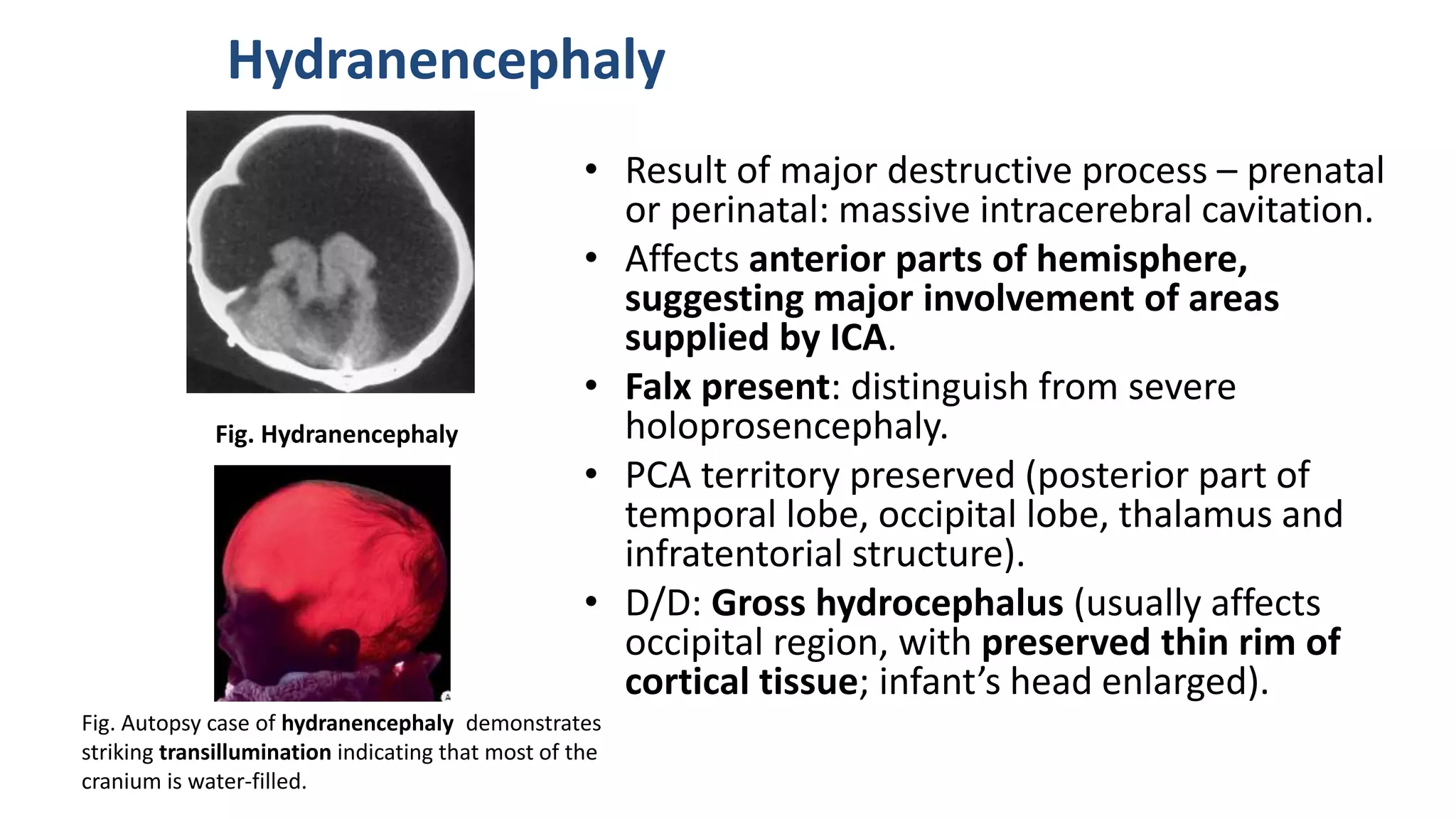


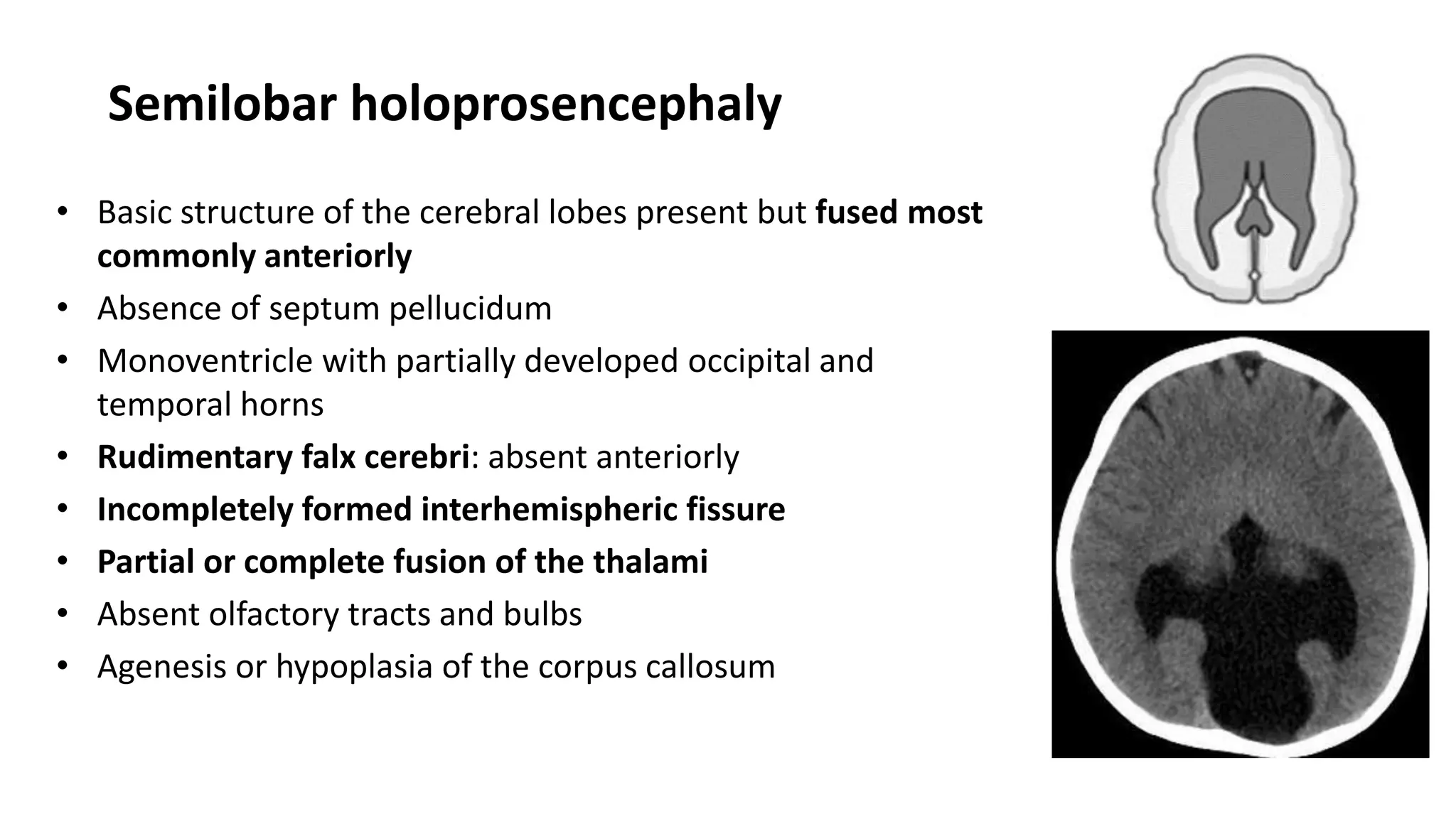

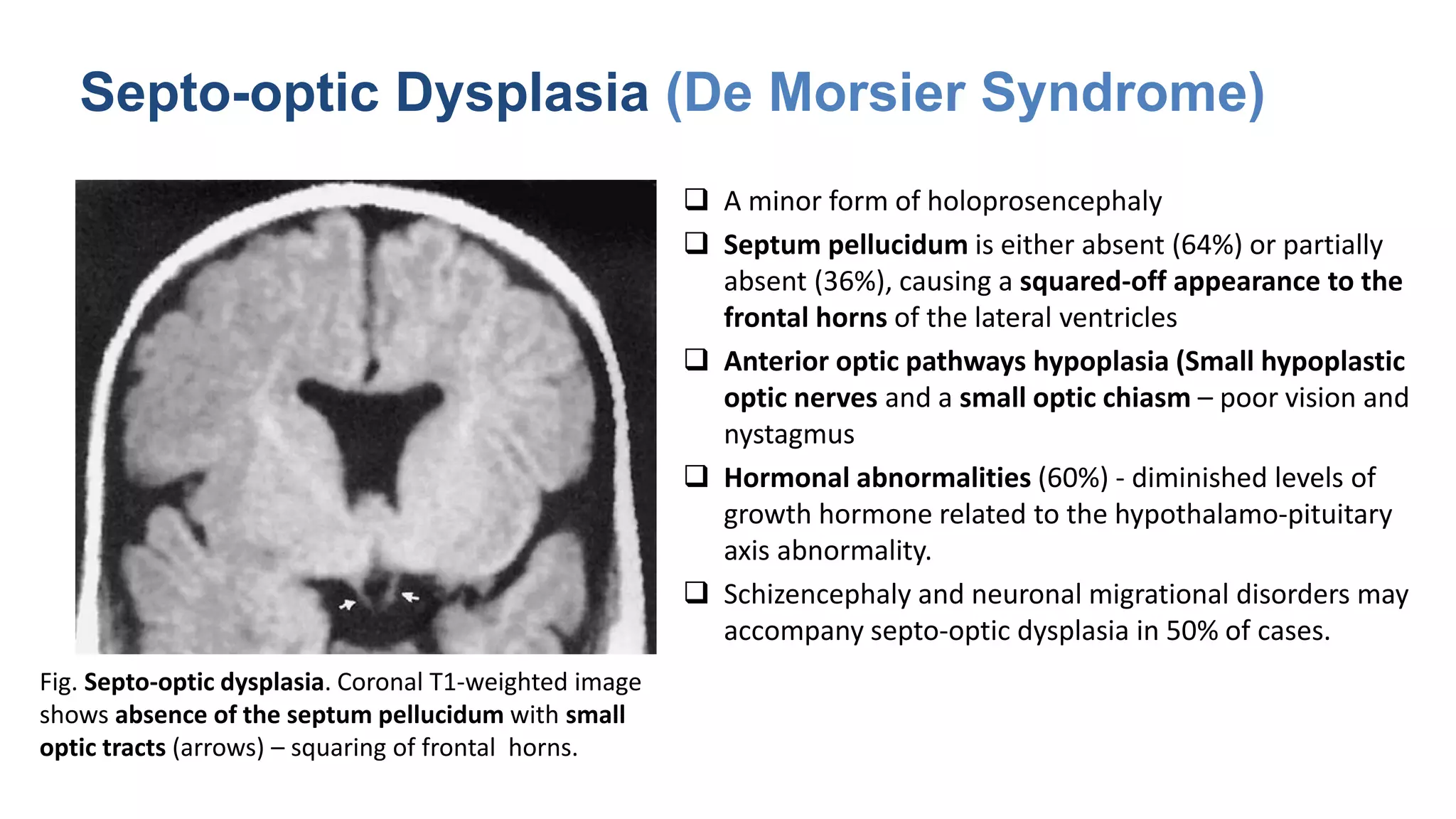






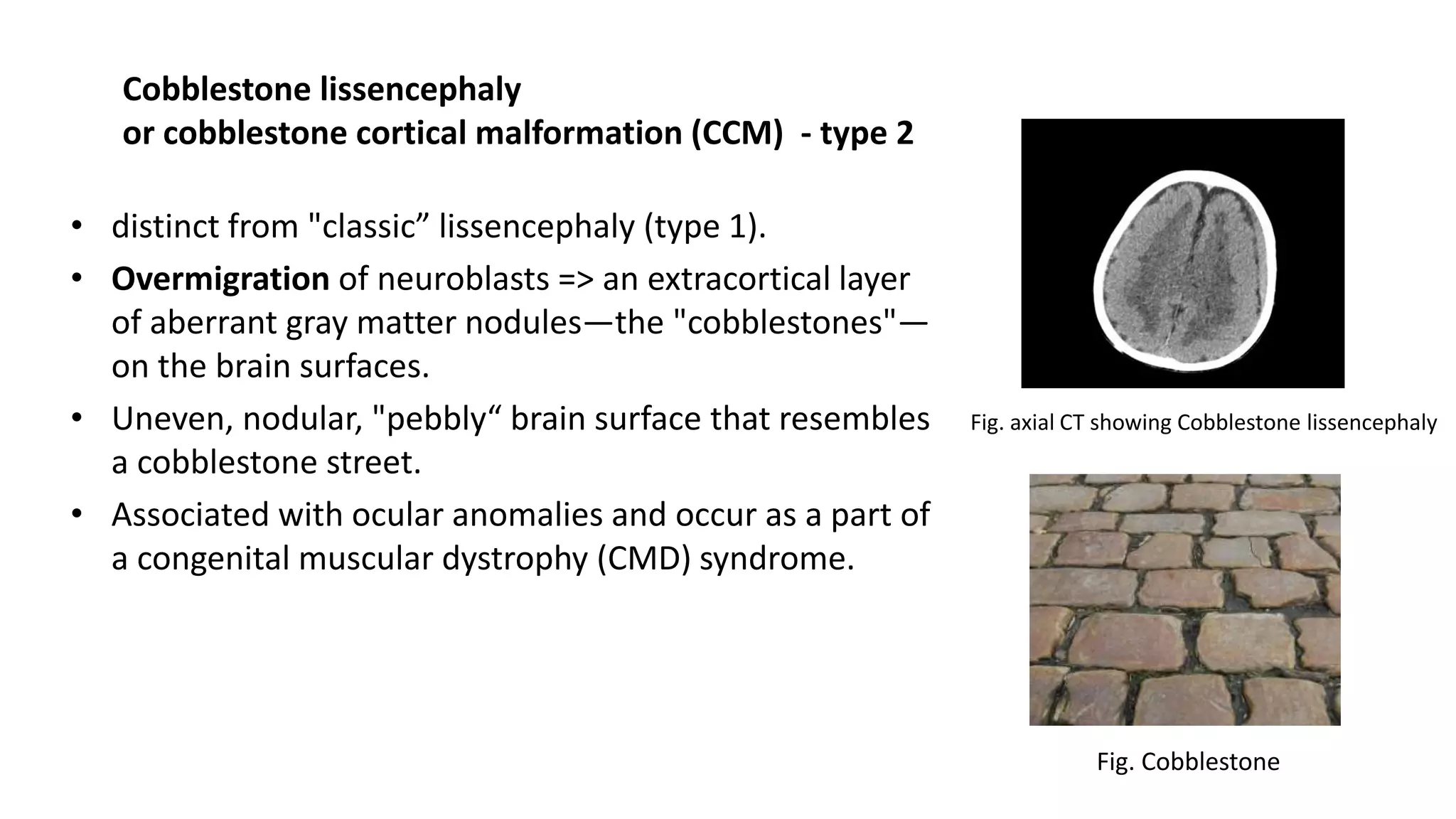





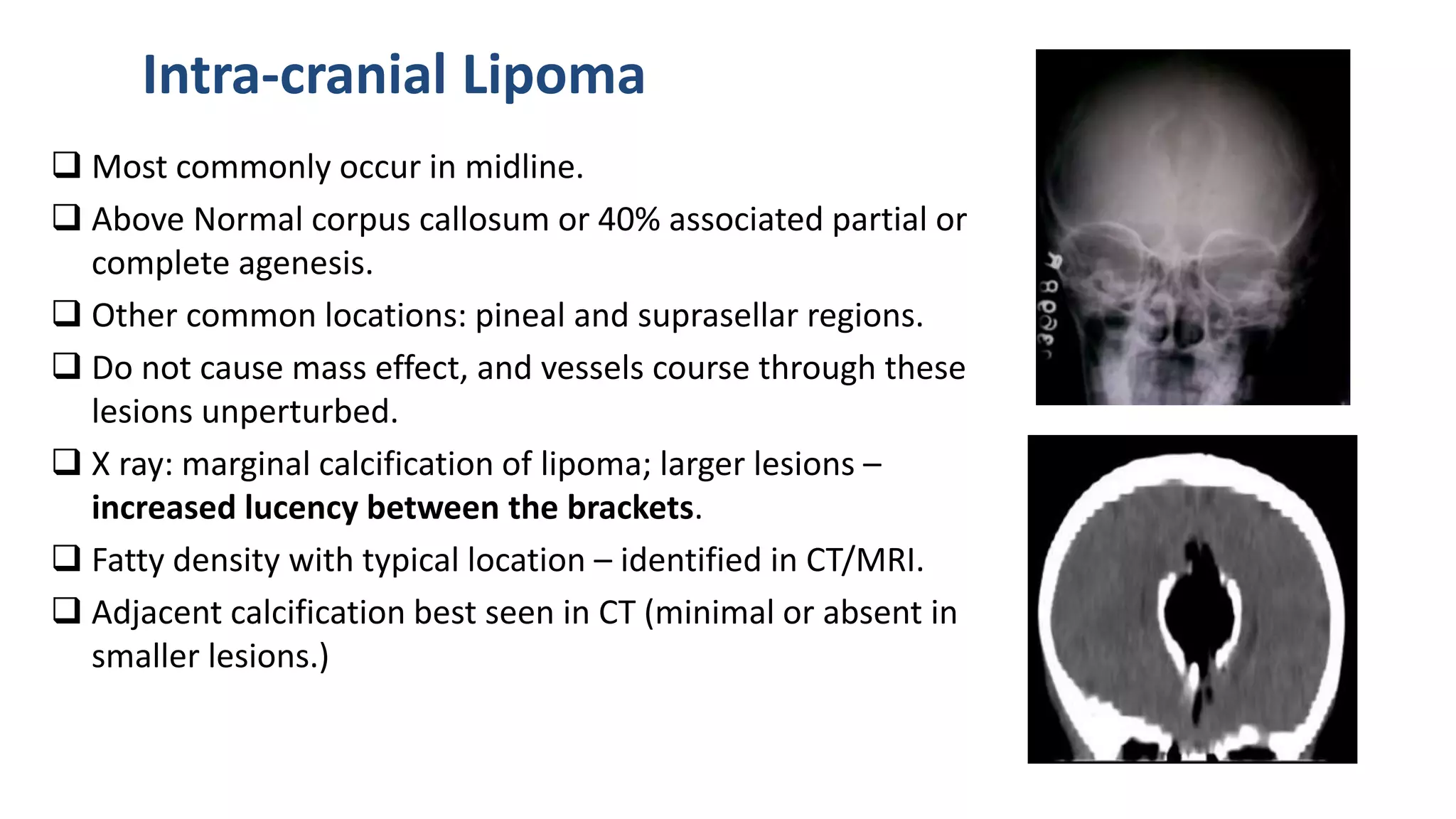



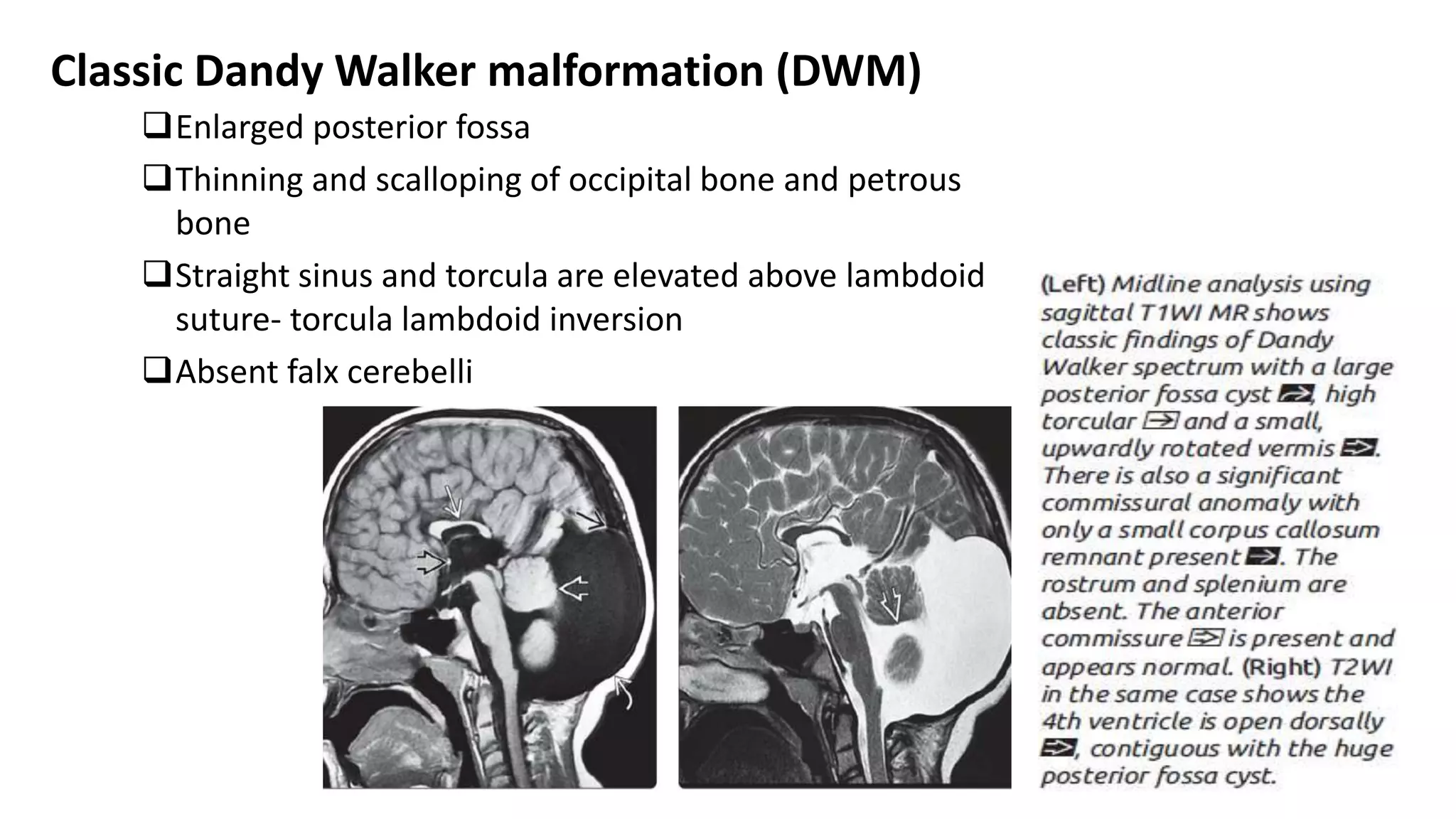
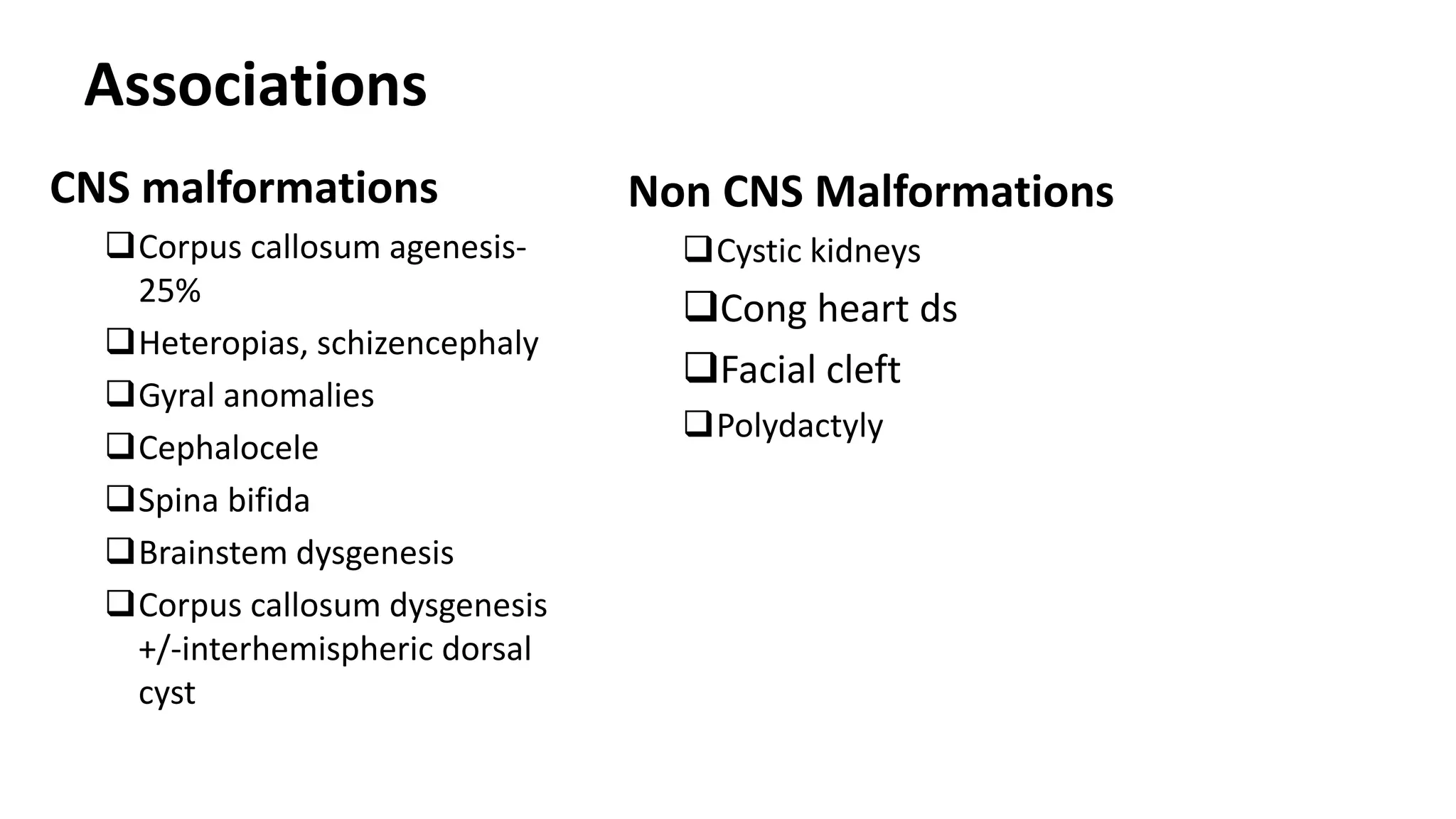

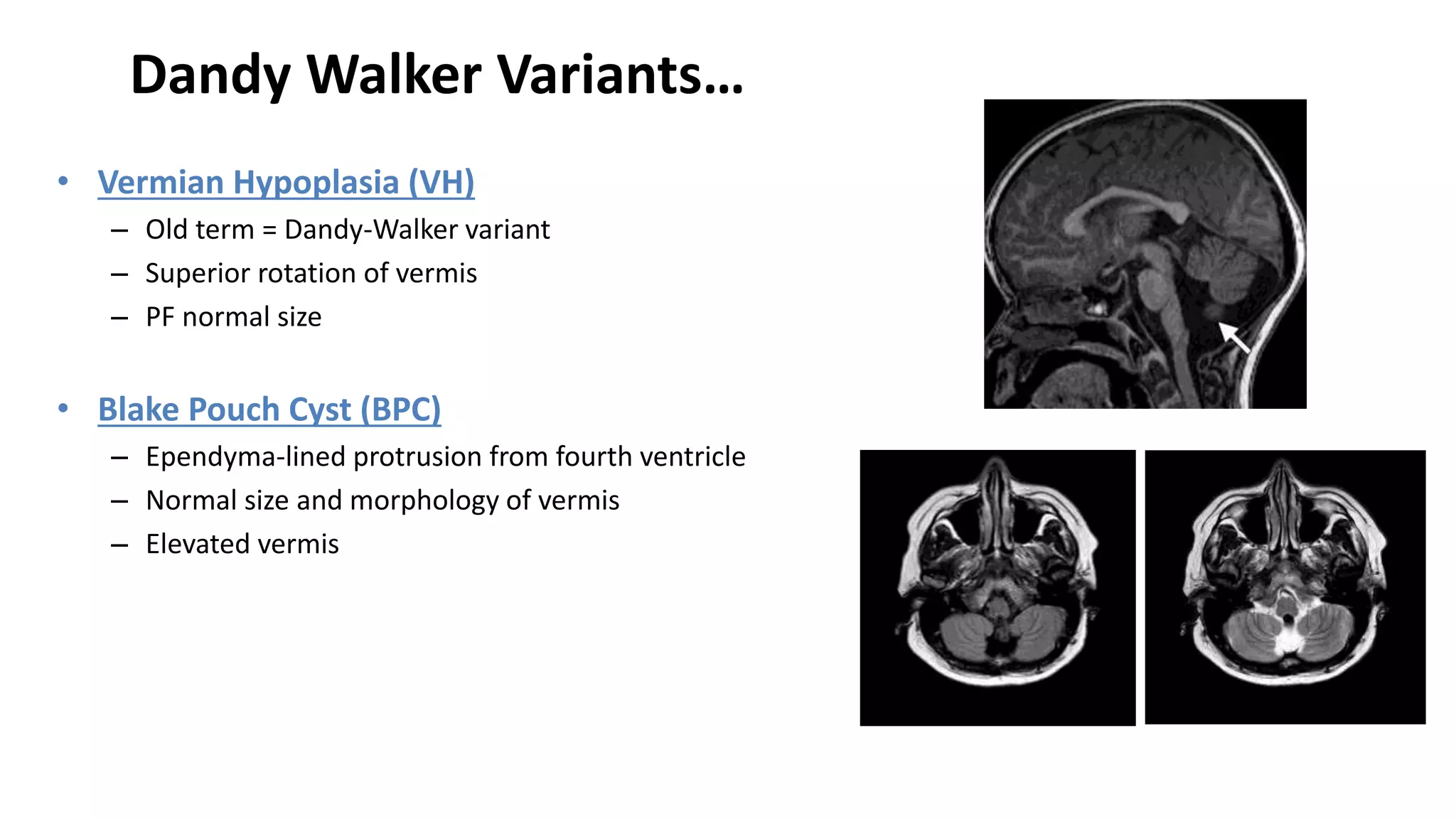







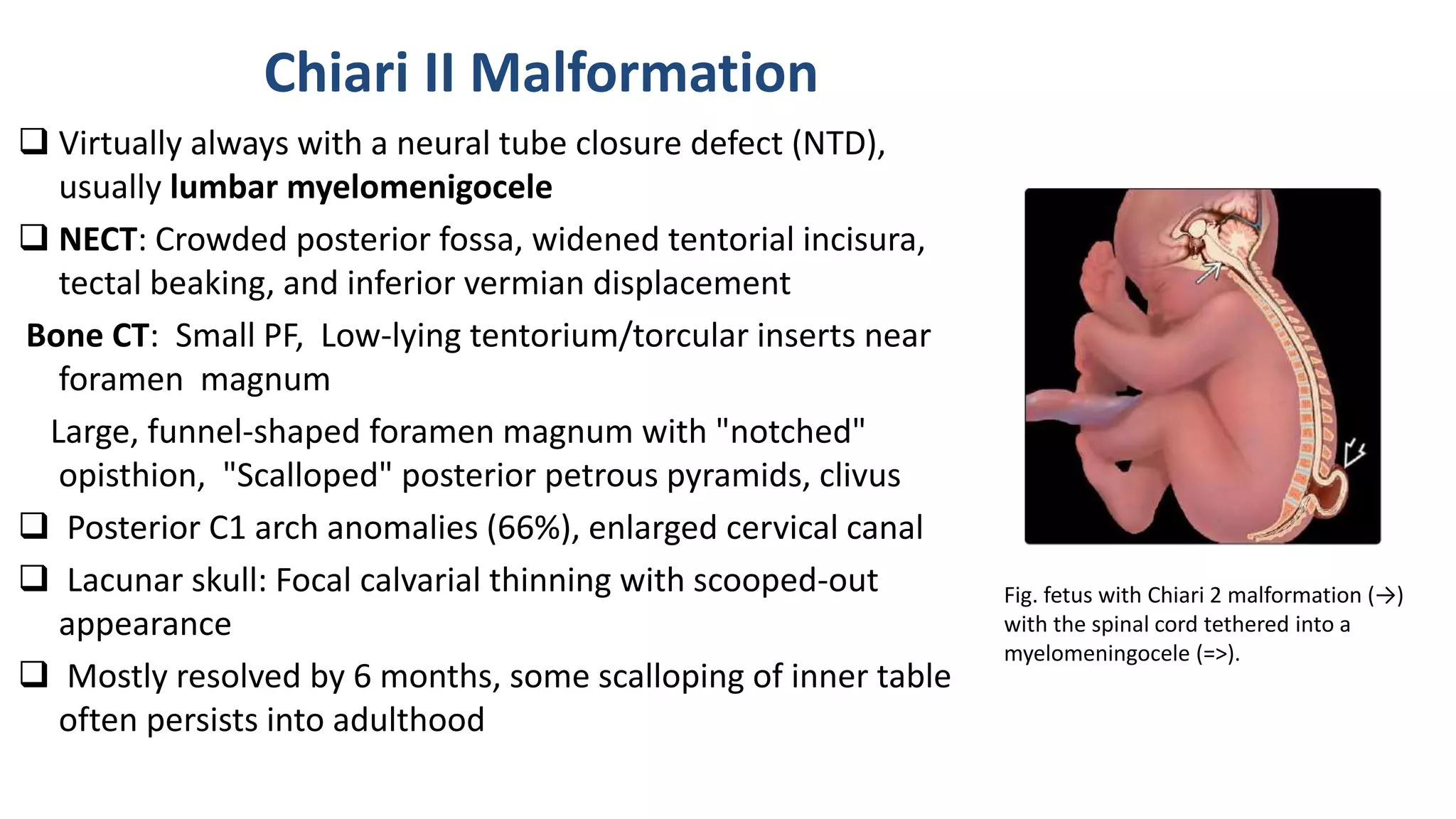


































This document discusses the normal development of the brain from embryology through maturation. It then reviews various congenital brain lesions that can occur due to disruptions during different stages of development including dorsal induction, ventral induction, neuronal proliferation and migration, and myelination. Specific lesions discussed include holoprosencephaly, septo-optic dysplasia, schizencephaly, corpus callosum agenesis, arachnoid cysts, and more. Imaging findings for each condition are also provided.























































































