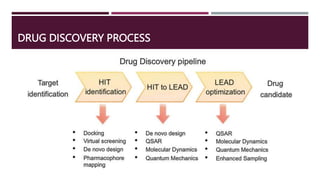
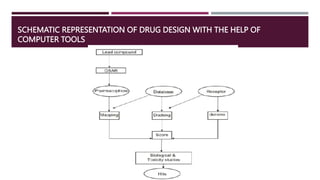
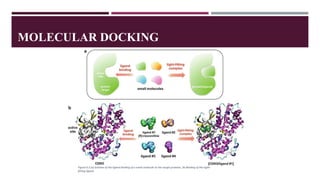
Computational chemistry plays an important role in drug design and discovery. The presentation outlines how computational methods can be used to identify hit compounds with activity against drug targets, improve lead compounds' potency, and optimize lead compounds into drug-like molecules. It discusses various computational approaches like molecular docking, molecular dynamics, and quantum mechanics that facilitate hit identification, lead generation and optimization. The goal is to computationally design safe and effective drug molecules that have qualities like bioavailability, metabolic stability, selectivity for target tissues, and minimal side effects.