










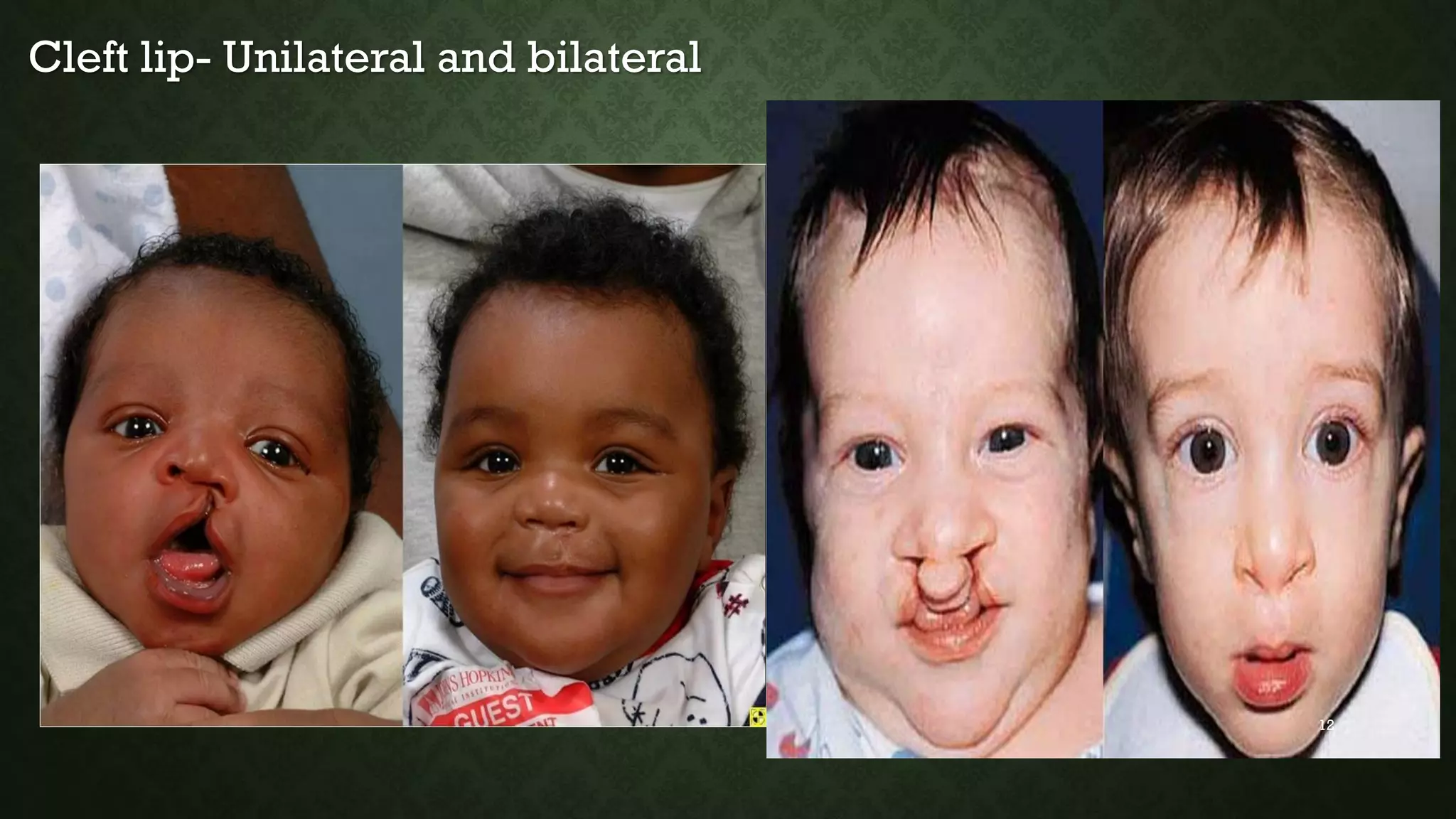














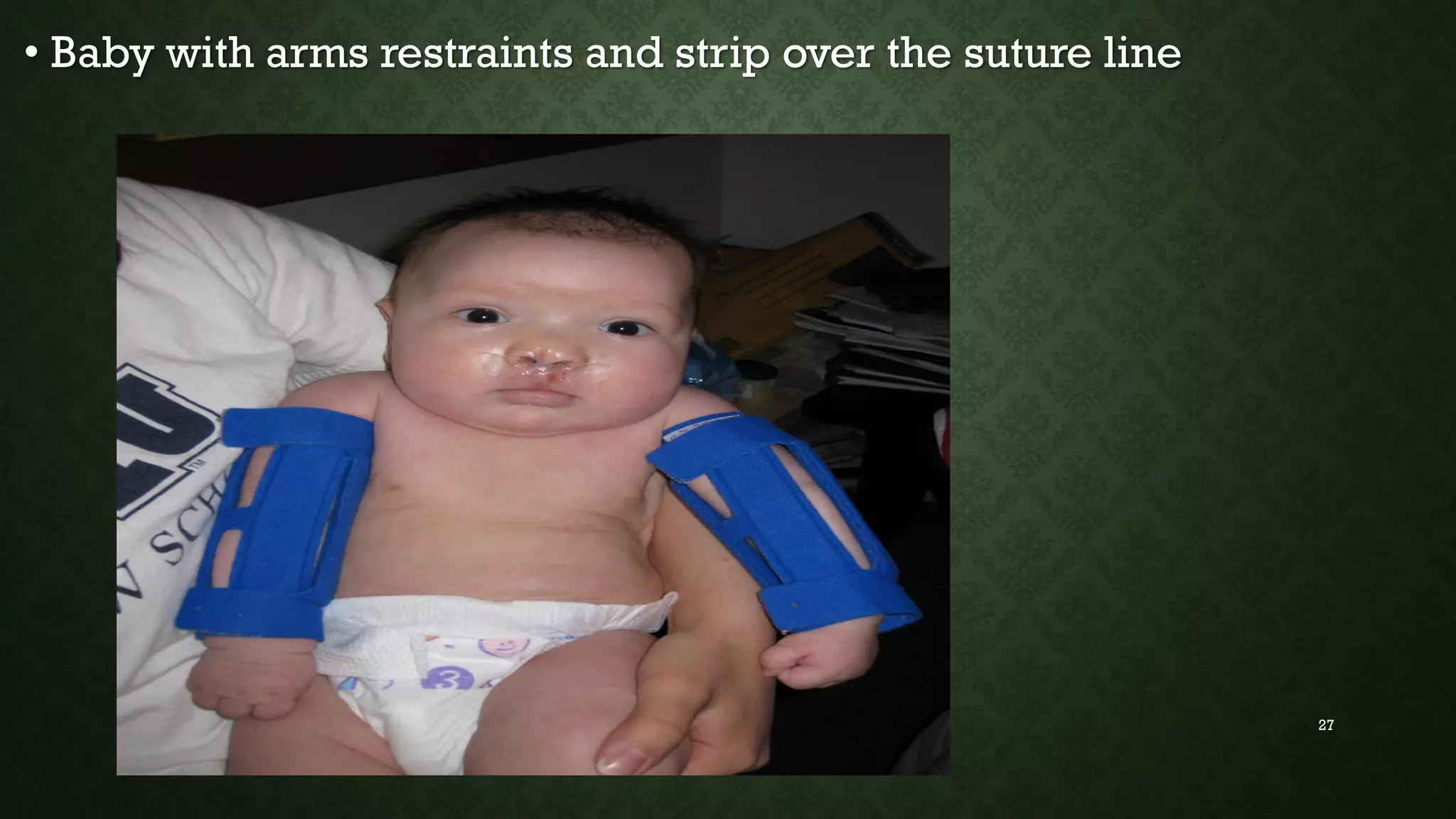



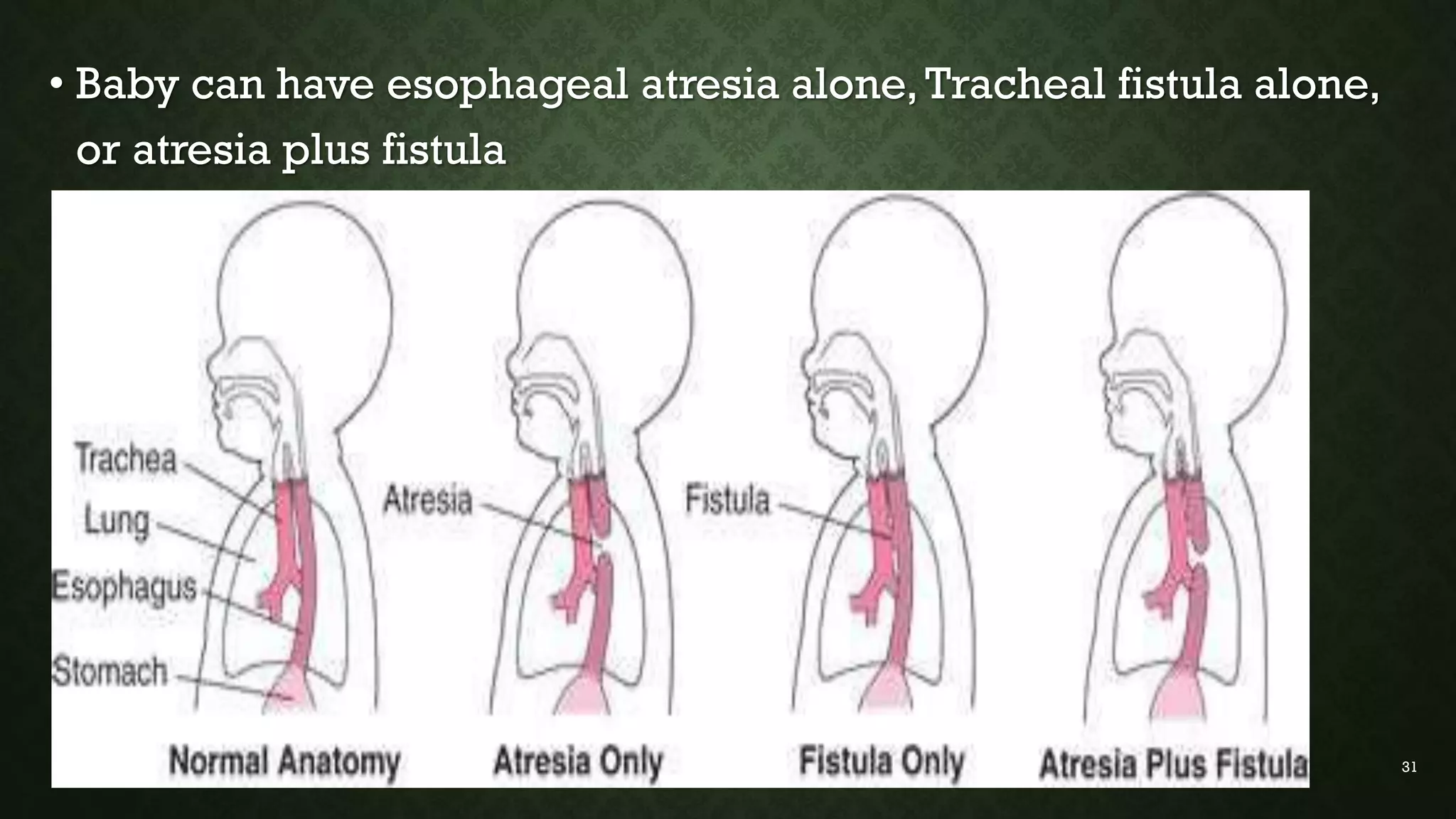







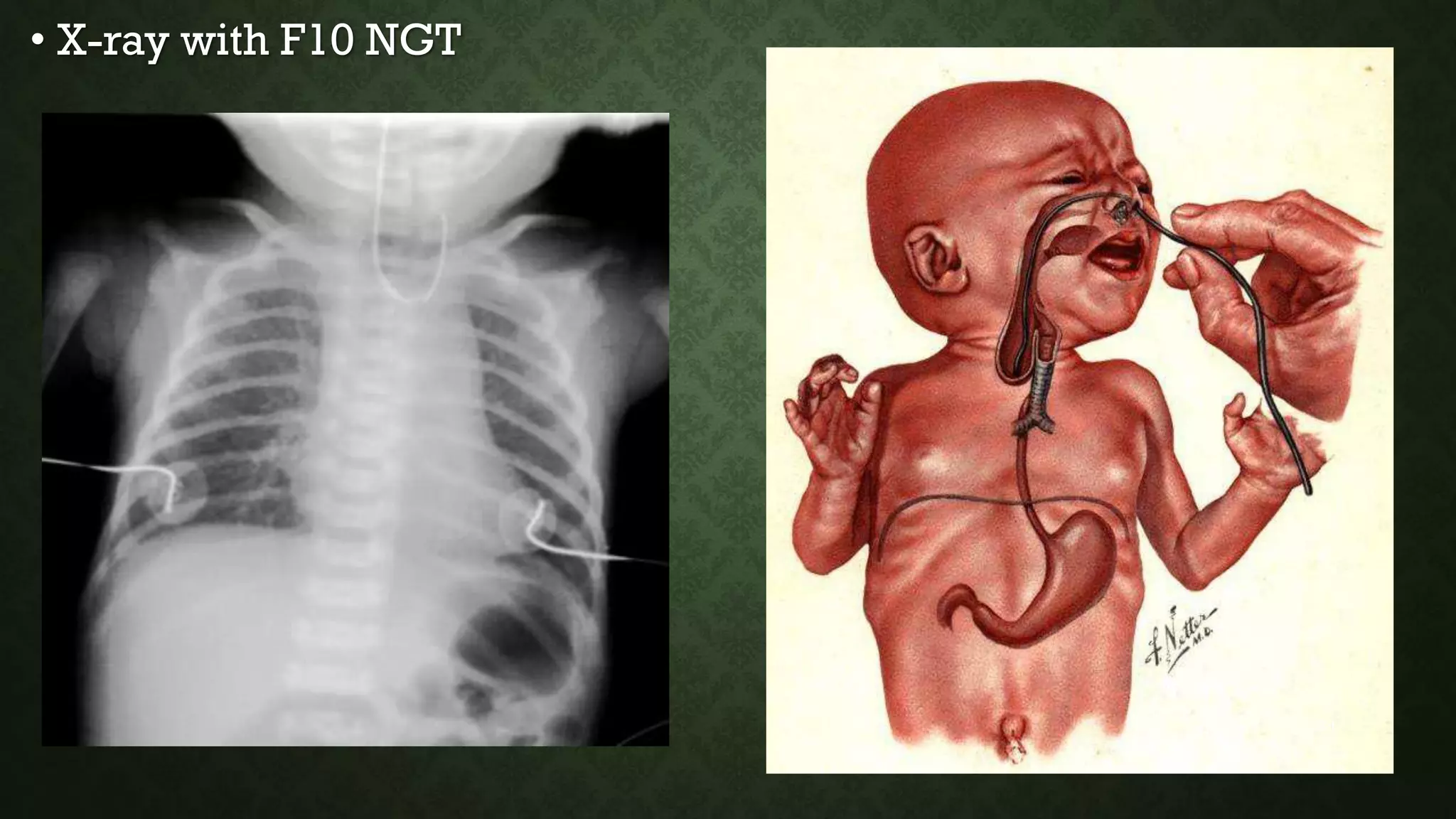






















This document discusses common pediatric surgical conditions, including gastrointestinal congenital anomalies present at birth. It identifies anomalies of the mouth (cleft lip and palate), esophagus (esophageal atresia and tracheoesophageal fistula), stomach/duodenum (pyloric stenosis), intestines (imperforate anus, omphalocele, gastroschisis, Hirschsprung's disease, intussusception), and provides descriptions, epidemiology, presentations, diagnoses, and management for each. It also outlines pre and post-operative nursing care for infants with surgical congenital anomalies, focusing on risks like infection, breathing issues, tissue trauma, nutrition and pain management.










































































