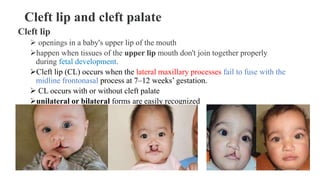
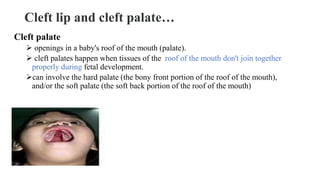
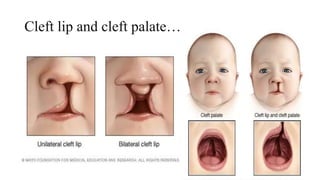
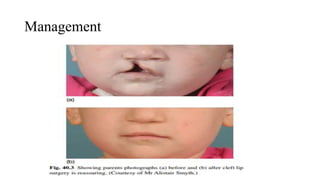
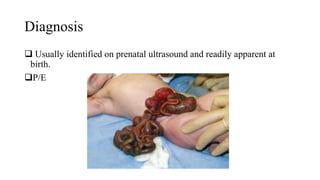
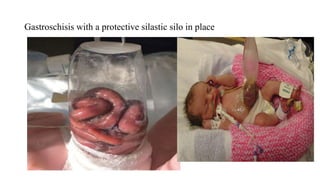
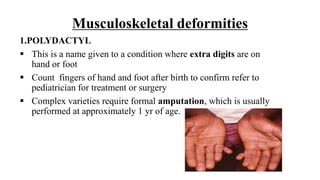
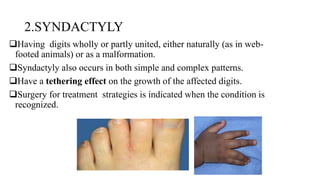
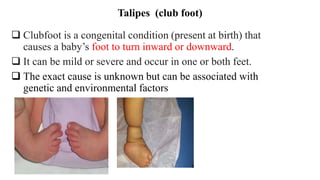
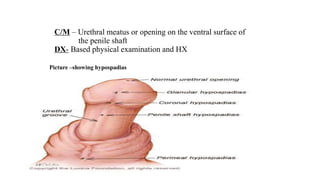
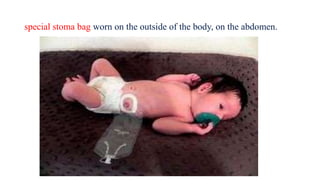
This document discusses cleft lip and cleft palate, including their causes, signs and symptoms, diagnosis, and management. Cleft lip occurs when tissues of the upper lip don't fuse properly, resulting in an opening. Cleft palate occurs when tissues of the roof of the mouth don't fuse, resulting in openings. Risk factors include family history and exposure to substances like smoking during pregnancy. Treatment may involve surgery to repair the lip and palate, as well as follow up care like speech therapy. Complications can include feeding and ear problems if not treated properly.