








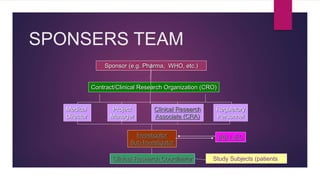

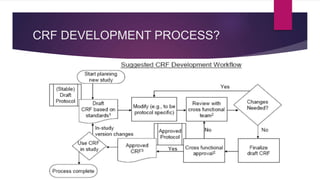




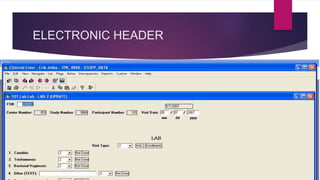


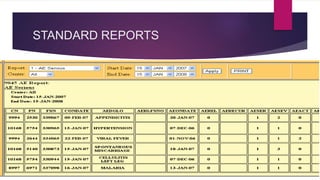











The Case Report Form (CRF) is a critical document in clinical trials, designed to capture protocol-required information for each trial subject, facilitating data collection, management, and analysis. It can be in paper or electronic form, with electronic CRFs offering advantages like real-time data handling and improved security. Guidelines for CRF completion emphasize accuracy, consistency, and proper management to ensure compliance and effective monitoring of trial data.



































































