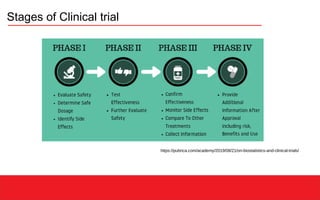
This document discusses clinical data management (CDM) which aims to ensure valid study data through collection, integration, and quality control of data. CDM involves developing data management plans, standard operating procedures, electronic case report forms, and using clinical data management systems. Key roles in CDM include clinical data managers who oversee data collection, validation, and storage in compliance with regulations. International guidelines like ICH-GCP provide principles for ethical and scientific conduct of clinical trials.






![Clinical data management systems
Tools for clinical trial data management, It ensures human errors are at the minimum by employing
multiple modes of varification
Typographic errors fixing
Logical errors (approve/disapprove if a condition is met)
Coding of data – Adverse event terms and medication names
●
Dictionary for adverse event – MedDRA, WHOART
●
Dictionary for medication --COSTART and WHO Drug Dictionary
These may suport IVRS facility for data collection
Perceptive Informatics, Medidata RAVE, Castor EDC,Forte Research Systems' OnCore eClinical, Aetiol EDC
[Jade Global Solutions {JGS}] and IBM Watson Health's IBM Clinical Development – Web Based for eCRF
XNAT LORIS, and EEGBase include support for imaging, electrophysiology, and other data modalities less
suitable to eCRFs](https://image.slidesharecdn.com/clinicaldatamanagement-200707140632/85/Clinical-data-management-7-320.jpg)