This document discusses clinical data management (CDM) systems and processes. It defines key terms like source data, source documents, and raw data. It then describes the essential steps in CDM including initial planning, data collection, review and verification, coding, query resolution, data entry and validation, output and archiving. Finally, it outlines requirements for a good CDM system including system validation, security, change control, and archiving. The goal of CDM is to generate an accurate, high-quality clinical trial database while ensuring compliance with regulations.
Introduction to Clinical Data Management (CDM) as a critical process in clinical research.
Definition and importance of source data and source documents in clinical trials including records from various clinical observations.Types of source documents which include originals and automated records used in clinical investigations.
Importance of good data management systems and the involvement of CROs in improving clinical trial data accuracy.
Objective of GCDMP to ensure high-quality databases adhering to ICH guidelines and FDA regulations.
Initial planning and establishing standardized systems for tracking data effectively throughout clinical trials.
Importance of continuous incoming data accuracy and the system for timely clarification of data errors.
Role of the data review committee and error detection procedures essential during review and verification of data.
Description of various data entry approaches, verification methods and their significance in ensuring data quality.
Different coding standards for adverse events, concomitant diseases, and challenges faced in coding.Processes for handling and resolving data queries, including documentation, tracking, and audit trails.
Final steps in data management including review, freezing of data and responsibilities in archiving essential documents.
Requirements for acquiring and compiling source data, including electronic data challenges and verification.
Details on transferring data from paper to electronic formats and the necessity of training staff involved.
Overview of critical system requirements for maintaining a robust CDM framework ensuring compliance and data security.Summation of the significance of CDM and technological advances aiding improved data management practices.
SOURCE DATA
• RECORDS
•ORIGINAL RECORDS OF CLINICAL FINDINGS
• OBSERVATIONS IN CLINCAL TRIALS
• RECONSRTUCTION AND EVALUATION OF THE TRIAL
• SOURCE DATA ARE CONTAINED IN SOURCE
DOCUMENTS
3.
SOURCE DOCUMENTS
• ORIGINALDOCUMENTS,DATA,RECORDS
• HOSPITAL RECORDS,CLINICAL & OFFICE CHARTS
• LABORATORY NOTES & FINDINGS,MEMORANDA
• SUBJECT’S DIARIES OR EVALUATION CHECKLISTS
• PHARMACY DISPENSING RECORDS
• RECORDED DATA FROM AUTOMATEDINSTRUMENTS
• COPIES OR TRANSCRIPTIONS CERTIFIED AFTER
VERIFICATION AS BEING ACCURATE COPIES
4.
SOURCE DOCUMENTS
• MICROFICHES,PHOTOGRAPHICNEGATIVES
• MICROFILM OR MAGNETIC MEDIA
• X-RAYS
• SUBJECT FILES
• RECORD KEPT AT PHARMACY
• RECORDS AT THE LABORATORIES
• MEDICOTECHNICAL DEPARTMENTS INVOLVED
IN THE CLINICAL TRIAL
5.
Source Document: Theelectronic record to used
to keep together a collection of eSource data
items for capture, transmission, storage,
and/or display; and serving as a source
document for a clinical investigation.
Raw Data: Data as originally collected. Distinct
from derived. Raw Data includes original
observations, measurements and activities
6.
INTRODUCTION
• CRO’s
• DATAGENERATION & PRESENTATION
• ACCURACY OF TRAILS & REGULATORS
• INFORMATION TECHNOLOGY (IT)
COMPUTERIZED SYSTEM (REMOVAL OF
TRADITIONAL SYSTEM PAPER WASTAGE )
• GROWTH & REQUIREMENTS OF GOOD DATA
MANAGEMENT SYSTEMS THAT COMPANIES WHICH
ARE OTHERWISE IT-BASED
7.
• HAVE FULLFLEGED CLINICAL TRIAL DATA MANAGEMNET
SYSTEMS WHICH BRING THEM A GOOD AMOUNT OF
BUSINESS AND REVENUE
• CDM is a fundamental process which controls data
accuracy of each trial besides helping the timelessness to
be achieved.
• It helps in linking clinical research co-ordinator = who
monitor all the sites & collects the data
• Link with biostatisticians = who analyze, interpret and
report data in clinically meaningful way.
8.
Good Clinical DataManagement
Practice (GCDMP)
• The objective of GCDMP is to generate high quality
database devoid of errors and omissions
• ICH GUIDELINES
• US FDA REGULATIONS
DRUG AND DEVICE DEVELOPMENT PROCESS
The Society of Clinical Data Management (SCDM) has
created a comprehensive document- Good Clinical
Data Management Practices (GCDMP) (Version 4.0 is
the most recently updated version published in May
2007)- that provides guidance on accepted practices of
Clinical Data Management (CDM)
10.
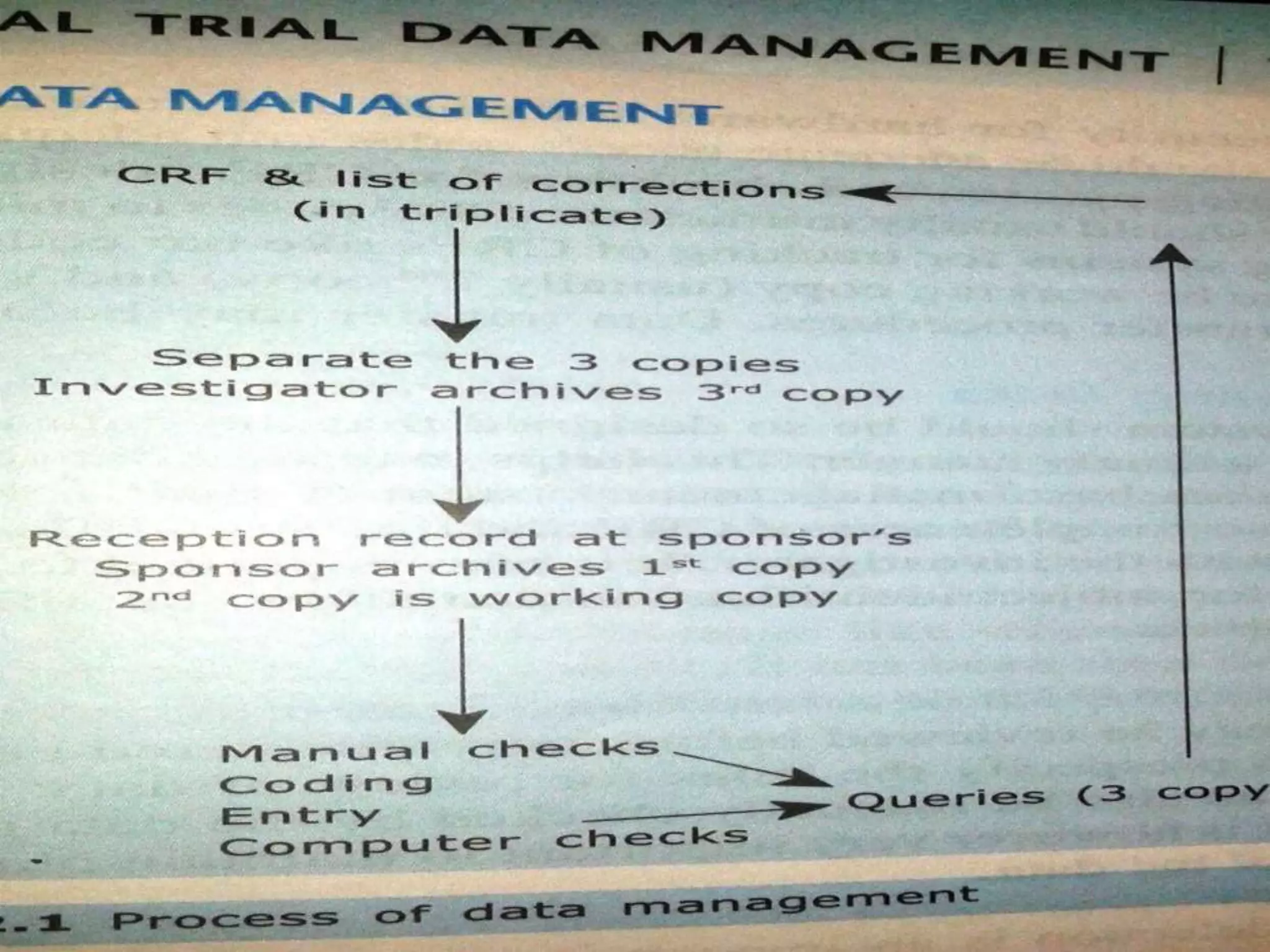
SYSTEMIC APPROACH FORCDM
• INITIAL PLANNING
SPONSOR or INVSTIGATOR or CRO
Standardized database management system
CRF CASE RECORD FORMAT
CRF as per database need, setting realistic
dates for receipt, verification, query
resolution, corrections, Final editing and
release of data and finally resource
mobilization
11.
• Preparing forIncoming Data Data
management study master file SOP’s should
be established to ensure operational
documentation for computers, system
reliability, Validation and accuracy.
• System security for hardware software and
data from theft and sabogate.
• Adequate access code and back up of the
data.
• Indexes & Checklists for CRF’s
• Designing data entry screens
12.
• Establishing systemsfor tracking of CRF;s like
Barcodes, deciding which CRF copy to be
working copy (usually second copy)
• Validating CRF and other data transfer
procedures.
• Data Transfer may be on Paper or Electronic
13.
INCOMING DATA
• Datareceived continuously and in a timely
manner
• Helps in data testing methodology, validates
data base management system (DBMS), helps in
checking accuracy and completeness of CRF
• Timely clarification of errors and omissions with
the investigators.
• It is also important to decide on unambiguous
Codes for subject identification that allow
identification of all the data of any subject.
14.
INITIAL DATA REVIEWAND
VERIFICATION
• DATA REVIEW COMMITTEE MEMBERS
• MAINTAINING BLINDING DURING REVIEW AND
ENTRY OF THE DATA
• ERROR DETECTION IS AN IMPORTANT STEP TO BE
DONE BEFORE AND DURING DATA REVIEW AND
VERIFICATION
• THE VARIOUS ERRORS THAT ONE CAN EXPECT
DURING THIS STAGE CAN RANGE FROM MISSING
DATA, FAULTY COMPLETION OF
FORMS,QUESTIONABLE VALUES (E.G. HEIGHT 20
FEETS), TREND TESTS TO GROSS PROTOCOL
VIOLATIONS
15.
• SUBSEQUENT ERRORSCAN ALSO BE DETECTED
AT VARIOUS STAGES LIKE DURING COMPUTER
ENTRY, ERRONEOUS CODING OR
INVESTIGATOR’S CORRECTIONS NOT BEING
TAKEN INTO ACCOUNT.
• DATA MONITORING COMMITTEE HELPS IN
ASSESSING THE PROGRESS OF TRIALS AT
INTERVALS TO RECOMMEND WHETHER TO
CONTINUE, MODIFY OR STOP THE TRIALS.
• IT ALSO EVALUATES SAFETY DATA AND CRITICAL
EFFICIACY END POINTS.
• THERE SHOULD BE WRITTEN OPERATION
PROCEDURES AND MAINTENANACE OF ALL
MEETING RECORDS
16.
DATA ENTRY, VERIFICATIONAND
VALIDATION
• The Data entry person should be defined for the
specific trial & specified in a data management
plan.
• For transcription from paper CRF to electronic
CRF different procedures are used:
Double Data Entry form (one person)
Double Data Entry form (two persons)
Single entry with second look
Single data entry with reading aloud
Single data entry with source verification
17.
• Double dataentry is not required by regulation by
good practice.
• Data entry process should be chosen based on the
skills of the personnel, this will give good impact on
to the resources in the project and the reflected
evaluation of key variables.
• Only authorized persons should be entitled to do
entry and corrections on the data entry screens.
• Verification and Validation is done by Data
Reviewers, automated computer checks (an error
message like when a value is outside the acceptable
norms) and during audit
• It has been that errors in entry is 1 % by good
operator.
• This Decreases to 0.1% by double entry of data by
two different operators.
18.
CODING
• FOR AdverseEvents
COSTART (Coding Symbols for Thesaurus of
Adverse Reaction Terms)
WHO-ART (Adverse Reaction Terminology)
SNOMED (Systematized Nomenclature of
Medicine)
MedDRA (Medical Dictionary for Regulatory
Activities)
In House Codes
19.
• FOR concomitantdiseases: international
classification of diseases version 10 (ICD-10)
• FOR concomitant medications: WHO Drug
Dictionary
• Medical Term ----- Preferred term(s)----- Code
ERRORS IN CODING
• Misunderstanding about medical terms,
misinterpretation of hand writing, defective
translation, foreign Language of CRF, wrong
choice of preferred terms and difficulties in
transcoding.
This errors leads to inconsistencies in
final report, decreased credibility of report,
delay in report writing and represent evidence
of negligence
20.
• To minimizeerrors only qualified and trained
staffs should be employed in the process the
data entry operators should insist on legible
filling of CRFs.
• It can also be minimized by keeping a log
book of difficult coding cases, doing
translation-retranslation and centralizing of
the final coding
21.
DATA QUERIES
Problems facedby data entry operators
• Subject has to go back to investigator
• Operators are failure to check the inclusion and exclusion criteria
• Inconsistent Calendar Dates
• Illegible entries
• Unfamiliar Drugs Names
• Text in unfamiliar Language
• Entries in incorrect place at CRFs
• Failure to specify indication for concomitant medication
• Lack of reason for change in medication
• Inconsistencies in physical examination at start and finish
• Incomplete information on Adverse Events
• Varying Units & Normal ranges in case of Laboratory Data.
22.
Query Tracking andResolution
• A proper SOP has to be made in place of query
tracking and solving
• Operator should draw a list of QUERIES
• This List should be sent to investigators who
verifies, corrects, signs and corrects the dates
the query
• Three copies should be send to the same format
then
• To the data entry operator who operates the
same
23.
• At theend a validation program is done and
run to follow the program and check the
editing done.
• Any change or correction must be readily
spottable and is called as AUDIT TRIAL.
• This Trial may be given in the computers
where computers saves the date and time of
correction, new value along with old value
and access code used to make changes or on
paper
24.
DATA OUTPUT, REVIEW& FREEZING
• As the data comes the manager and
stastician finalizes the data and queries are
resolved.
• Thereafter a final audit is performed, data is
frozen and sent to the statistician.
• Goal of perfectly accurate database is usually
unrealistic.
• It is preferable to set acceptable limits of
error that do not alter the validity of
statistical analysis and results and
conclusions drawn from the study
25.
ARCHIVING
• Data mangersand statistician are responsible
for archiving the electronic database,
associated computer programs, Data
monitoring conventions, audit trials and final
report.
• They also maintain also all sponsor-specific
essential documents as per regulatory
requirements.
26.
PATIENT DATA
BUDGET/BILLING PHARMACEUTICAL
PROTOCOLCLINICAL RESEARCH ASSOCIATE
TRAINING TRIAL MONITOR
REGULATORY FORMS CONTRACT RESEARCH
RESEARCH SITE MANAGER /
INVESTIGATOR
CLINCAL DATA MANAGEMENT
SOLUTION GATHERS AND CENTRALIZES
DATA
27.
REQUIREMENTS FOR ACQUIRING/CAPTURING/
COPYING SOURCE DATA
• In general data & documents containing source data
must first be specified in the trial protocol.
• Source Data are the original data, the recordings
and all information regarding Clinical Investigations,
Laboratory findings, anamnesis, interviews, patient
diaries and other sources.
• The original documents have to be archived.
• Copies have to be dated and signed by a
responsible person (Certified copies)
• If the original data is stored electronically, a
printout has to be made or a list of dates and
versions of stored documents signed/dated by
Principal Investigator.
28.
• In thecase of eSource data, of course, this is
not possible.
• A copy of eSource data shall be accepted in
place of eSource data, if the copy hass been
produced and verified against the eSource
data based on procedures defined in a SOP
for acquiring data duplication and
verification.
• Appropriate handling is also required for
scanning source documents.
• The Scanning process has to be validated
prior to implementation in a trial to ensure
the integrity of the generated record
29.
• In theCRF is the source document (e.g., in
psychiatric instruments like psychometric
scales ) this has to be defined in the protocol.
• If work has been used as a transcription
instrument (e.g., Transitional documentation
prior to electronic data entry), these are to
be considered as informal source data sheets
and have to be filed and quality checked
appropriately.
• In general, source data must be accessible
and verifiable and the quality of digitisation
must be carefully controlled using
appropriately defined SOPs.
30.
pCRF to eCRFTransfer
• In this scenario, clinical data are at first collected
with a pCRF.
• Investigator has less time or has to move
between locations (e.g. emergency ward,
operation theatre)
• In a remote data entry scenario, it is often not
the investigator, but special assistance personnel
who enters data from the pCRF into the eCRF
• This transcription step must be quality assured.
31.
• Type ofpersonnel needed (i.e., for data
entry, for data review, etc.)
• Criteria chosen to qualify them must be
clearly defined.
• For using eCRF, specific training programs for
investigators and assistance personnel must
be included.
• Appropriate quality control steps have to be
implemented and double data entry may be
prformed.
• pCRF transfer as well as status (arrived, re-
viewed, non-correct, requested queries,
correct, closed) must be clearly tracked.
32.
• Personnel responsiblefor different phases of
pCRF entry must be traked as well as all the
changes.
• Because the investigator’s signature is
required, he is responsible for the correct
transcription of the data.
• Appropriate workflow support should be
implemented in the Electronic Data Capture
(EDC) system.
33.
ESSENTIAL REQUIREMENTS
GOOD CDMSYSTEM
• System evaluation and provider/vendor
selection.
• System installation, setup and configuration.
• System configuration management
(Configuration of Audit Trial e.g. reson for
change optional or not?).
• System access and profile management.
34.
• Change Control
1.Risk Assessment of any change in the system.
2. Controlled processes of making changes to the
system, consisting of announcement,
assessment and approval of the change.
• System Security
1. Password policy.
2. Firewall configuration.
3. Physical & Logical security, in particular also at
the sites (EDC).
4. System controls.
5. Network security for remote access.
35.
• Database andcommunication security
1. Encryption of data storage, data Transfer.
2. Electronic signature has to comply also with
national regulations [EDC].
• Data protection
1. Handling of personally identifiable data (e.g.,
blinding of additionally submitted identifying
data; sites should eliminate personal identifiers
from source documents prior to submission).
2. Specification of minimum subject identifiers.
3. Safeguarding that (future) use of data is in
accordance with informed consent.
36.
4 Regulation ofaccess to electronic or paper
based data storage.
5 Particularly strict standards for genetic data.
6 Secure data handling procedures.
7 Use of pseudonyms/anonyms where
appropriate.
8 Secure cross-border data transfer.
• Data backup and recovery
• Disaster system recovery
• Database security
37.
• Data Archiving
1.Database specification.
2. Data files.
3. Audit Trial.
4. Clinical Data (open standards – vendor
independent, e.g., CSV, XML, PDF, ODM,
from CDISC)
5. Archiving reports.
6. Scanned paper CRFs.
7. Content and Variable definitions (metadata).
38.
8 Report ondata completeness at respondent and
variable level.
9 Secure Storage and access control.
• Business continuity
• Migration of data/meta-data (in case of system
retirement)
• System Validation.
• Risk management.
1. All components of the system have to be judged
according to their risk to violate GCP.
2. GCP-compliance has to be guaranteed especially for
high-risk components.
3. Maintenance of GCP-compliance even after updates
or other changes to the system.
39.
• Periodic review/audits.
•Safeguard of Blinding.
• Help Desk.
CONCLUSION
The importance of CDM can be realized from
the fact a lot of pure IT companies are
involved in CDM activities and this
contributes a big share in their revenue.
Some of the advances in CDM are:
40.
• New hardware'slike PCs, Electronic
notebooks
• Remote data entry.
• Optical mark recognition like bar codes.
• Optical character recognition like
fingerprints.
• Facsimile.
• Smart cards for each patient.
• Computer assisted new drug application
[CANDA] by FDA.

































![• Database and communication security
1. Encryption of data storage, data Transfer.
2. Electronic signature has to comply also with
national regulations [EDC].
• Data protection
1. Handling of personally identifiable data (e.g.,
blinding of additionally submitted identifying
data; sites should eliminate personal identifiers
from source documents prior to submission).
2. Specification of minimum subject identifiers.
3. Safeguarding that (future) use of data is in
accordance with informed consent.](https://image.slidesharecdn.com/clinicaldatamangementsk-181125144148/75/Clinical-data-management-35-2048.jpg)




![• New hardware's like PCs, Electronic
notebooks
• Remote data entry.
• Optical mark recognition like bar codes.
• Optical character recognition like
fingerprints.
• Facsimile.
• Smart cards for each patient.
• Computer assisted new drug application
[CANDA] by FDA.](https://image.slidesharecdn.com/clinicaldatamangementsk-181125144148/75/Clinical-data-management-40-2048.jpg)




















































































