Download to read offline

![CONTENT
Introduction
Factor affecting on biopharmaceutical classification system
Biopharmaceutical classification system [classes]
Application of biopharmaceutical classification system
Application of preformulation considerations in the development of
a. Solid
b. Liquid oral
c. Parenteral dosage forms Its impact on stability of dosage forms.
2](https://image.slidesharecdn.com/bcs-240801052111-139f1b59/85/BIOPHARMACEUTICAL-CLASSIFICATION-SYSTEM-BCS-2-320.jpg)




![DISSOLUTION
It is process in which solid substance solubilises in given solvent i.e mass
transfer from solid surface to liquid phase.
Using USP apparatus I at 100 rpm or USP apparatus II at 50 rpm .
Dissolution Media [900 ml],
1. 0.1 N HCl or simulated gastric fluid (pH 1.2) without enzyme.
2. pH 4.5 buffer & pH 6.8 buffer.
3. Simulated intestinal fluid without enzyme.
7](https://image.slidesharecdn.com/bcs-240801052111-139f1b59/85/BIOPHARMACEUTICAL-CLASSIFICATION-SYSTEM-BCS-7-320.jpg)
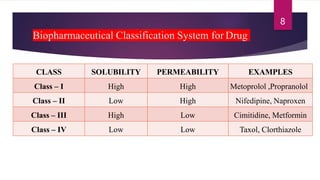














The document discusses the Biopharmaceutical Classification System (BCS), which classifies drug substances based on their aqueous solubility, intestinal permeability, and dissolution rate for predicting in vivo pharmacokinetics. It outlines the four classes of drugs (Class I to IV) based on these characteristics and their implications for drug absorption and formulation. The document also addresses the importance of stability studies and preformulation considerations in the development of dosage forms.















![Bio pharmaceutical classification System [BCS]](https://cdn.slidesharecdn.com/ss_thumbnails/biopharmaceuticalclassificationystembcs-160328061345-thumbnail.jpg?width=640&height=640&fit=bounds)


















































