





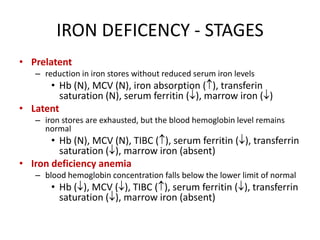
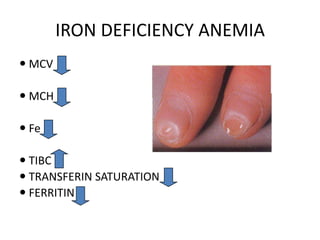













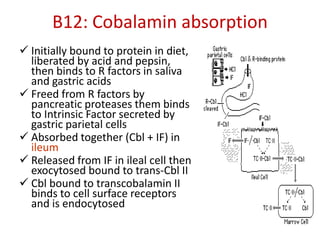




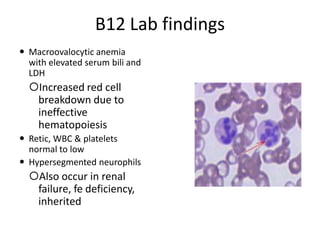
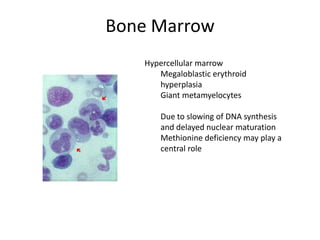
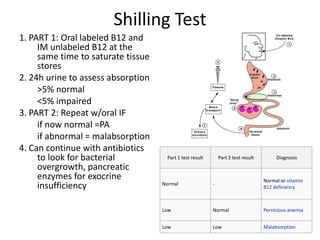





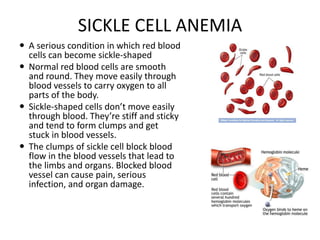
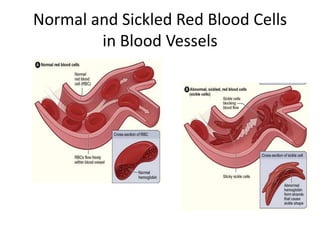


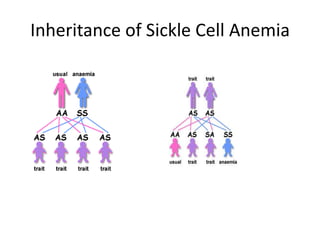







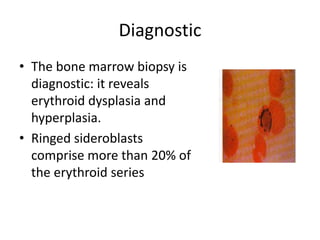





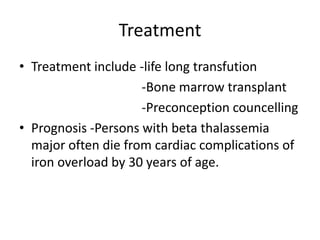


























This document provides an outline and summary of a seminar presentation on approaching patients with anemia. It discusses the definition and classification of anemia and covers several common types of anemia - iron deficiency anemia, anemia of chronic disease, megaloblastic anemias, sickle cell anemia, and thalassemia. For each type of anemia, the document outlines etiologies, symptoms, diagnostic criteria, bone marrow findings, and treatment approaches.



































































![ONFH[AVN HIP] -TRIPLE REGIME -A NOVAL SURGICAL CONCEPT .pptx](https://cdn.slidesharecdn.com/ss_thumbnails/onfhavnhip2026koaconcalicutdrgokuldevdrmashraf-260210064517-213ec005-thumbnail.jpg?width=640&height=640&fit=bounds)

















