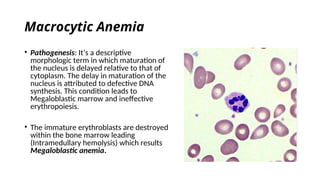
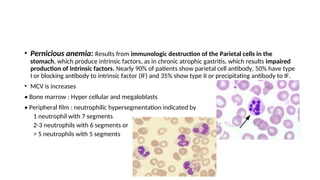
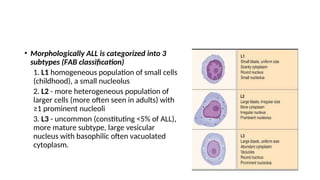
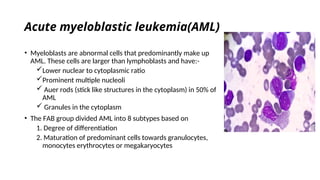
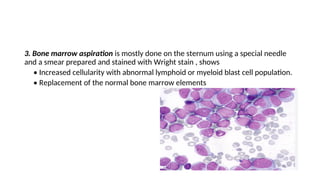
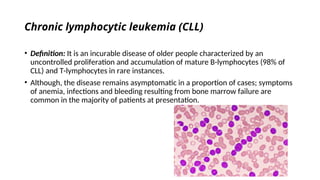
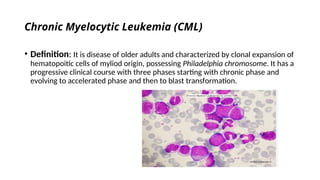
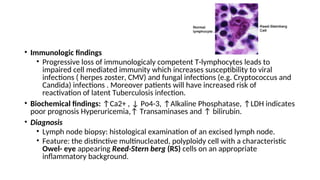
The document provides an extensive overview of hematologic disorders, focusing on anemia, its definitions, classifications, etiology, clinical manifestations, and diagnostic approaches. It discusses types of anemia including iron deficiency, anemia of chronic diseases, and macrocytic anemia, highlighting underlying causes and treatment strategies. Additionally, it covers leukemias, their classifications, epidemiology, and clinical presentations.