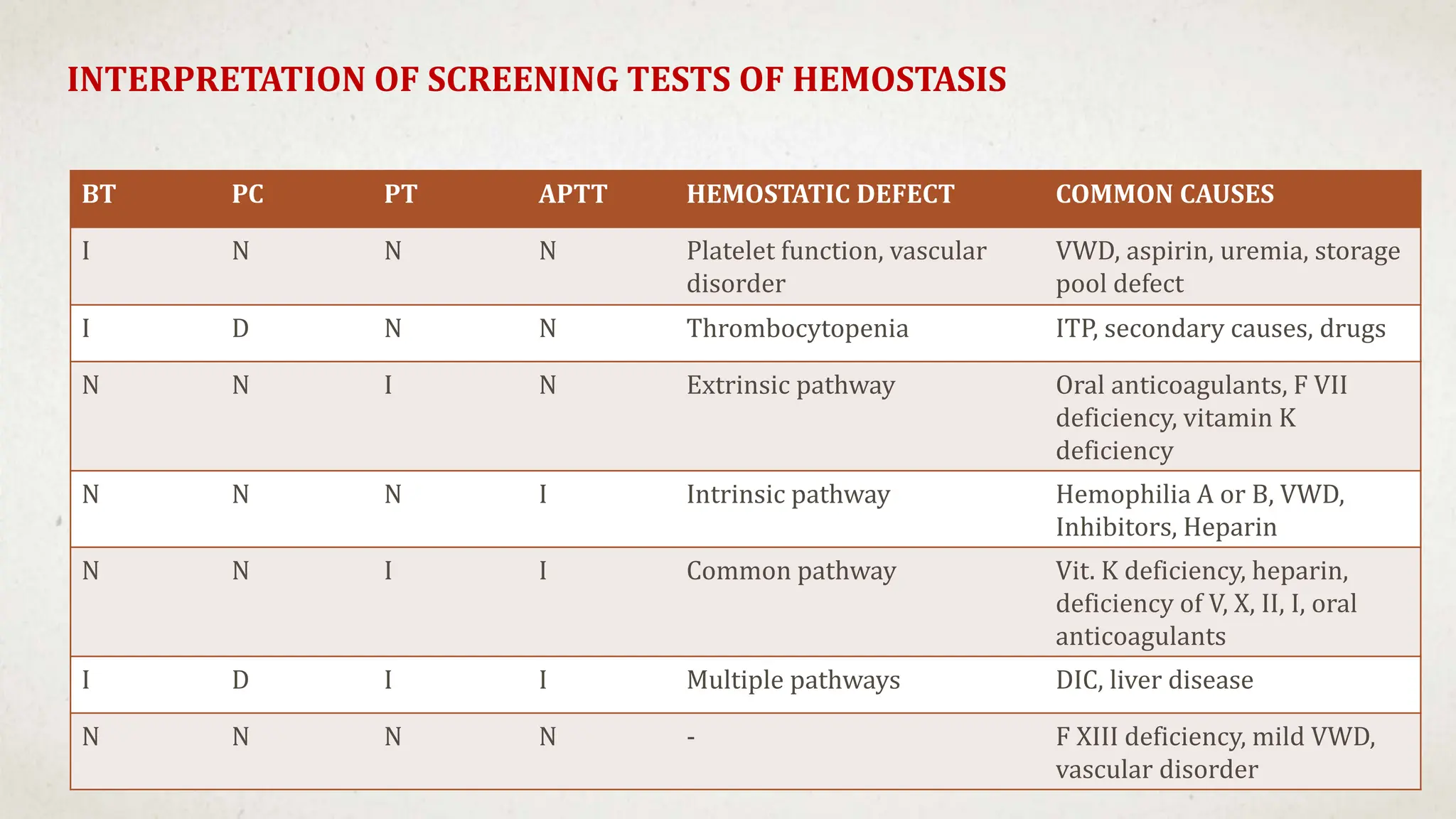
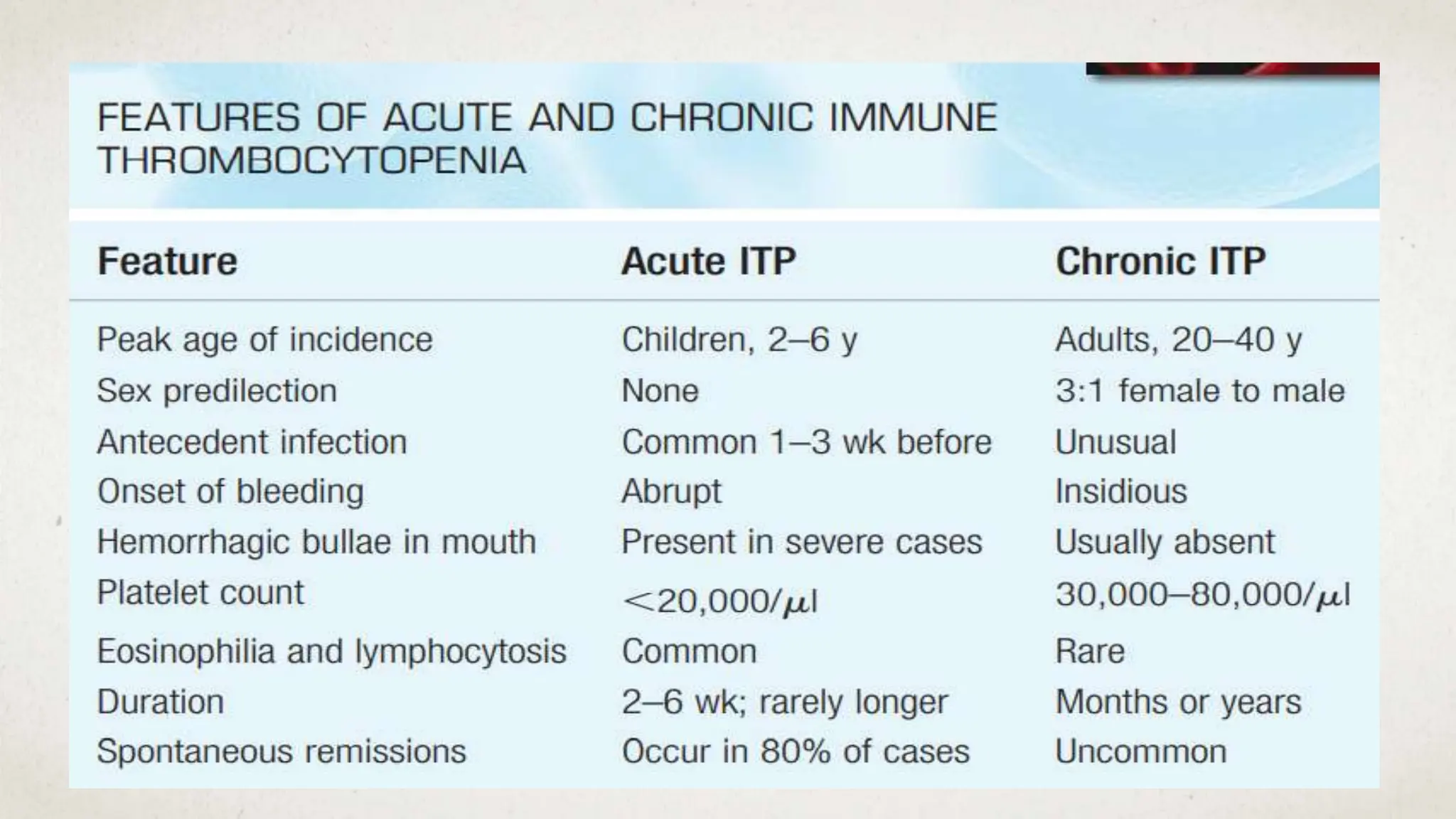
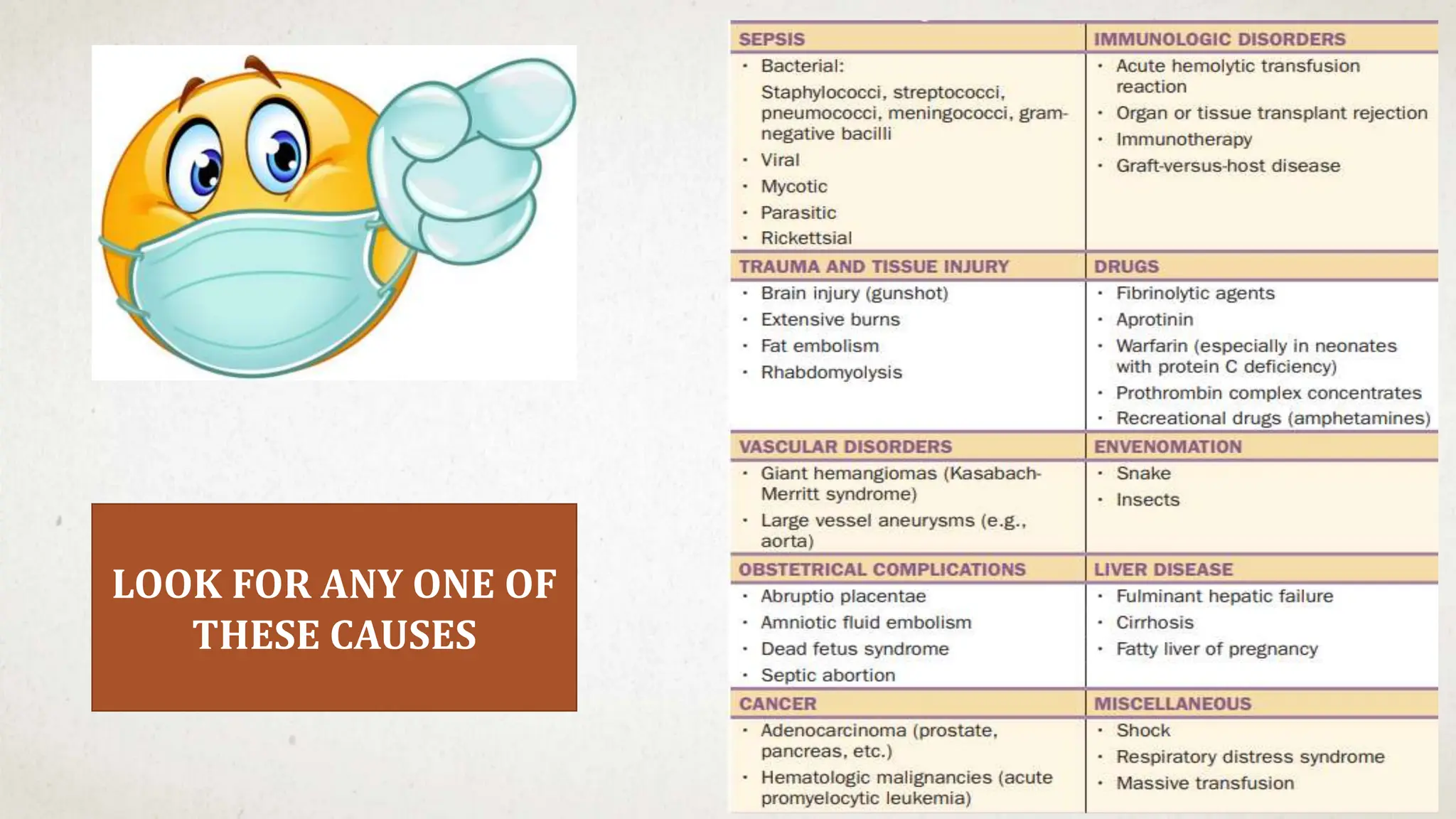
This case suggests a diagnosis of thrombocytopenia. The key features are:
- Recent history of infection suggesting a secondary cause
- Purpura and petechiae on examination
- No family history suggesting an acquired rather than inherited cause
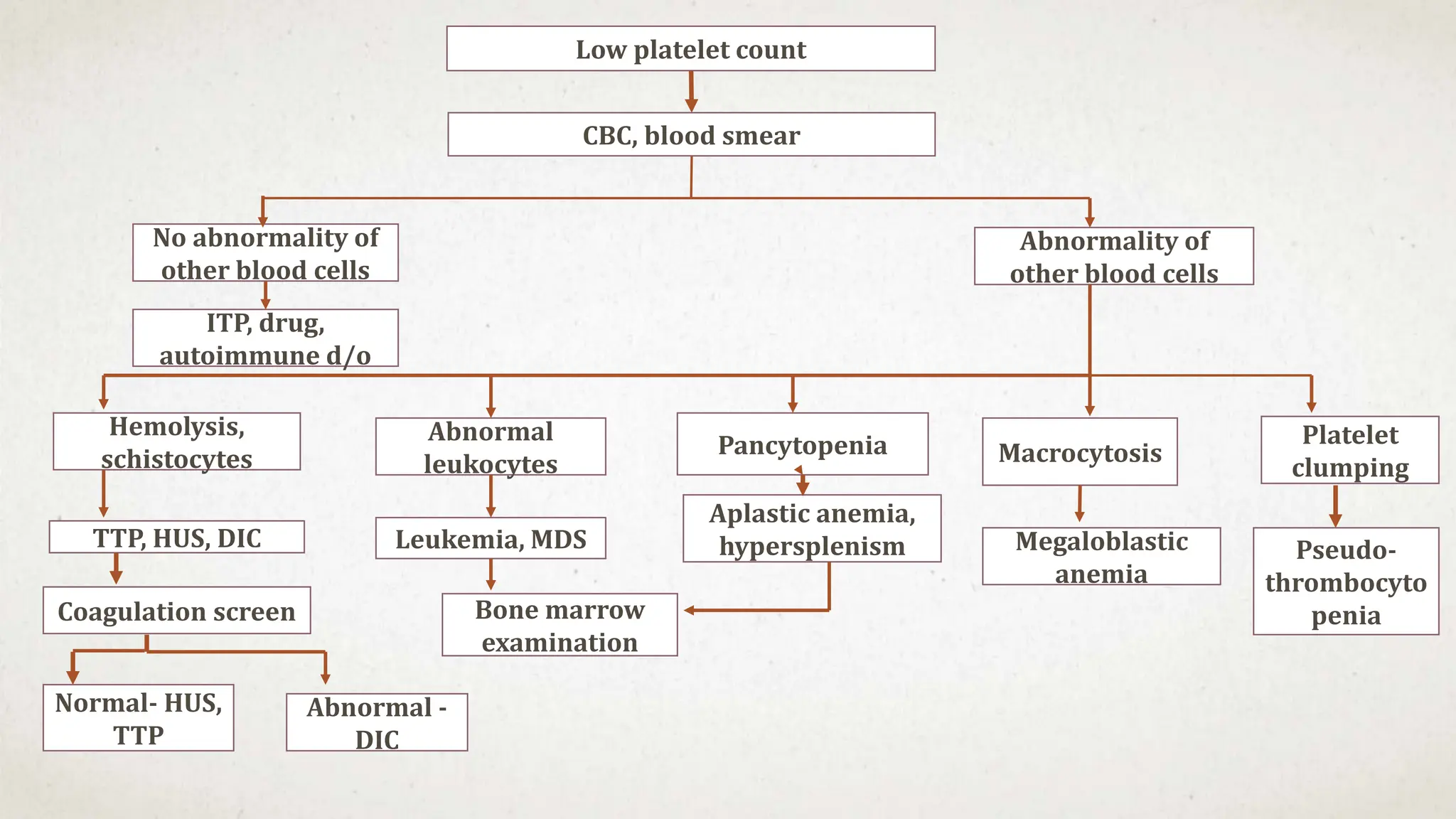
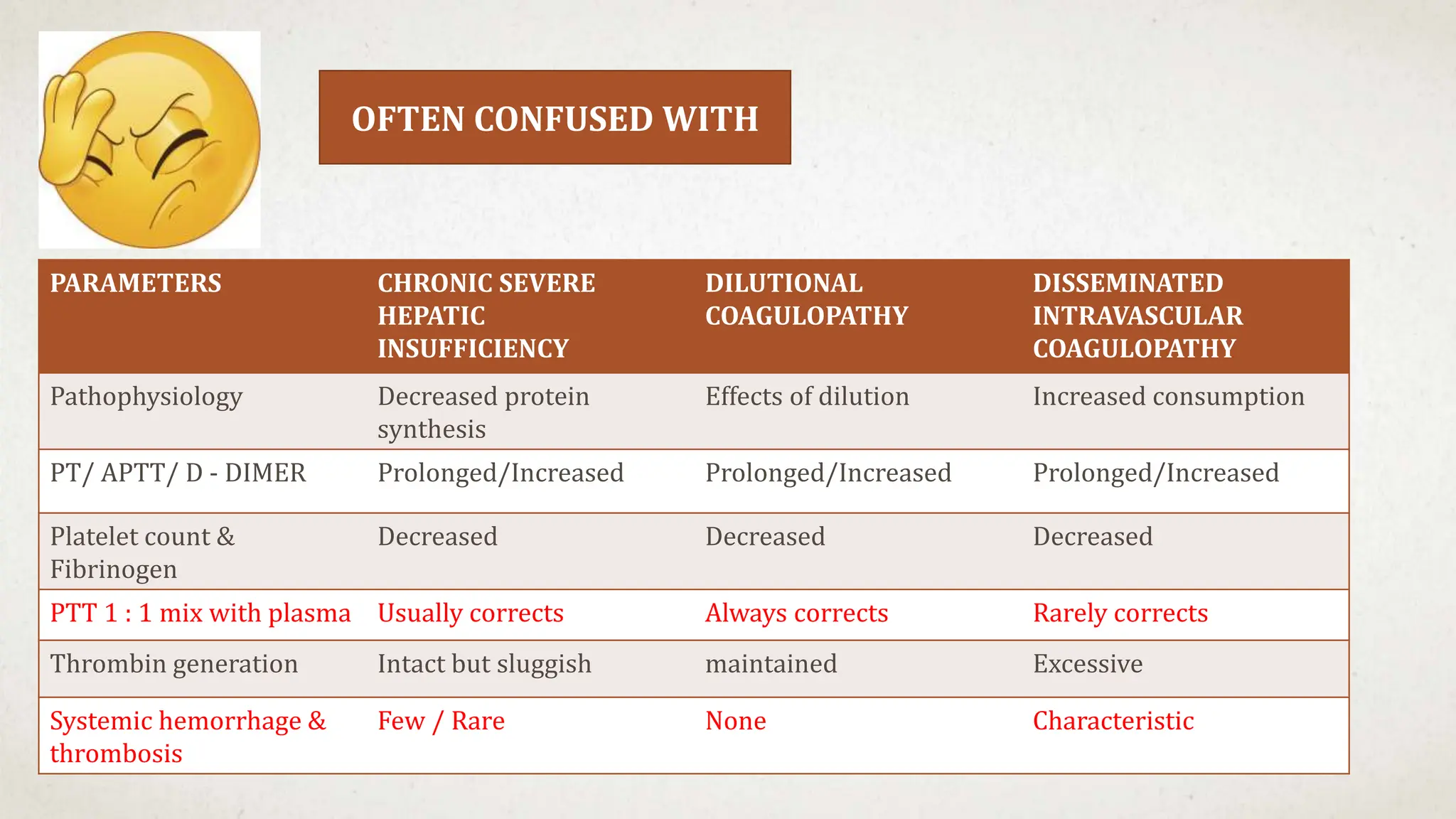
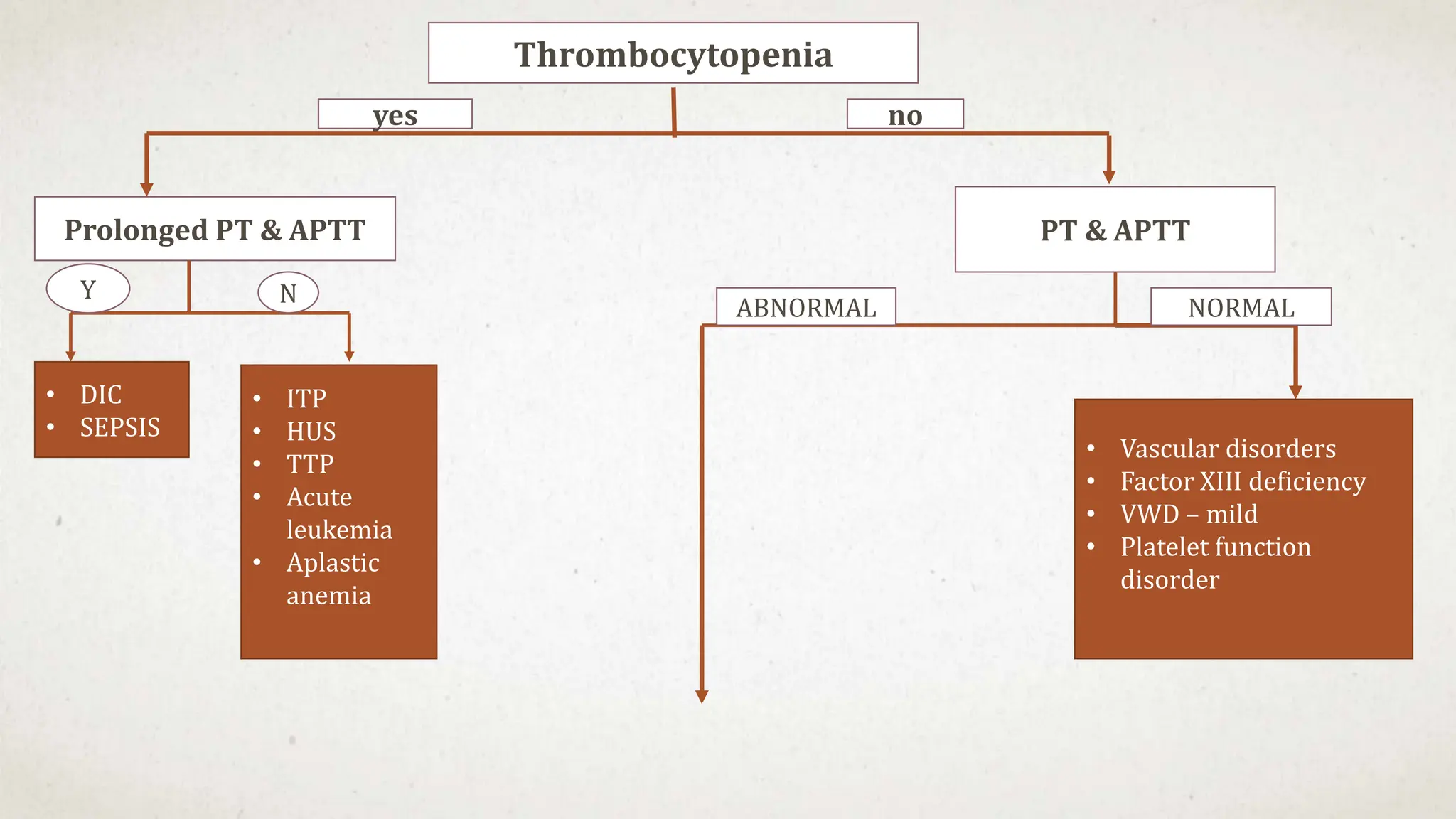
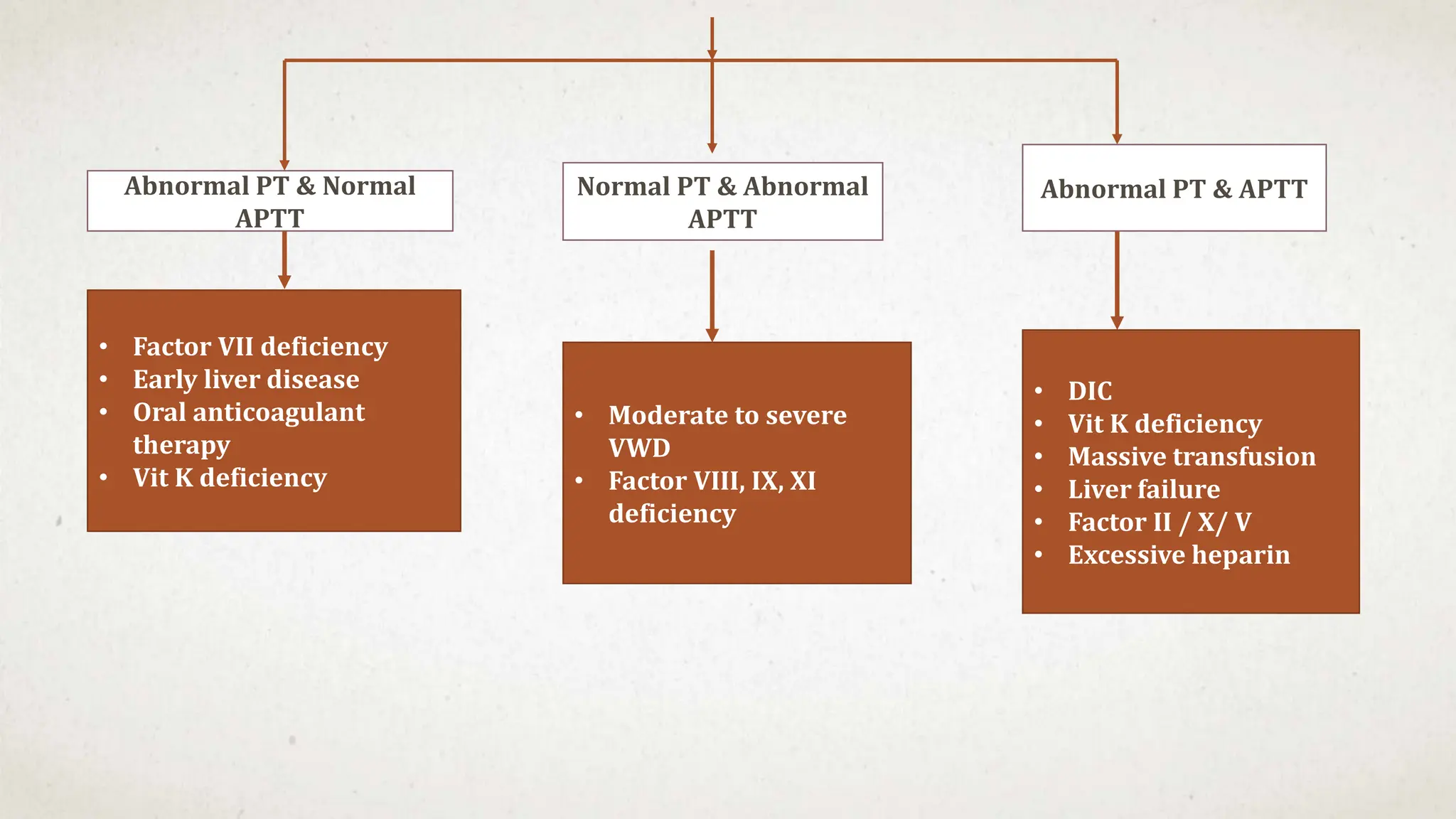
- Thrombocytopenia is a likely diagnosis which would explain the bleeding manifestations.
Further workup should include a CBC with platelet count to confirm thrombocytopenia and evaluate for possible secondary causes like ITP.




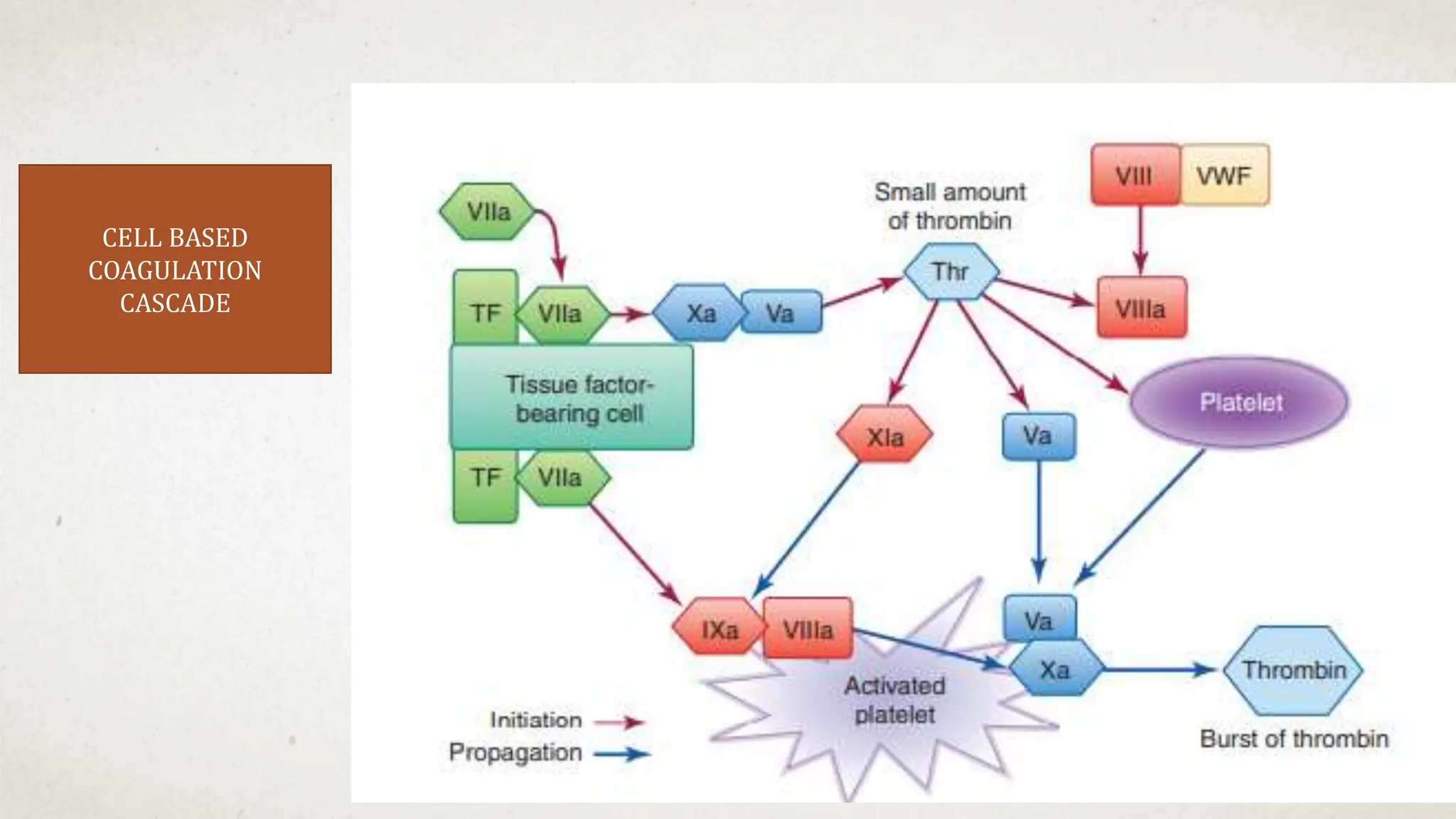
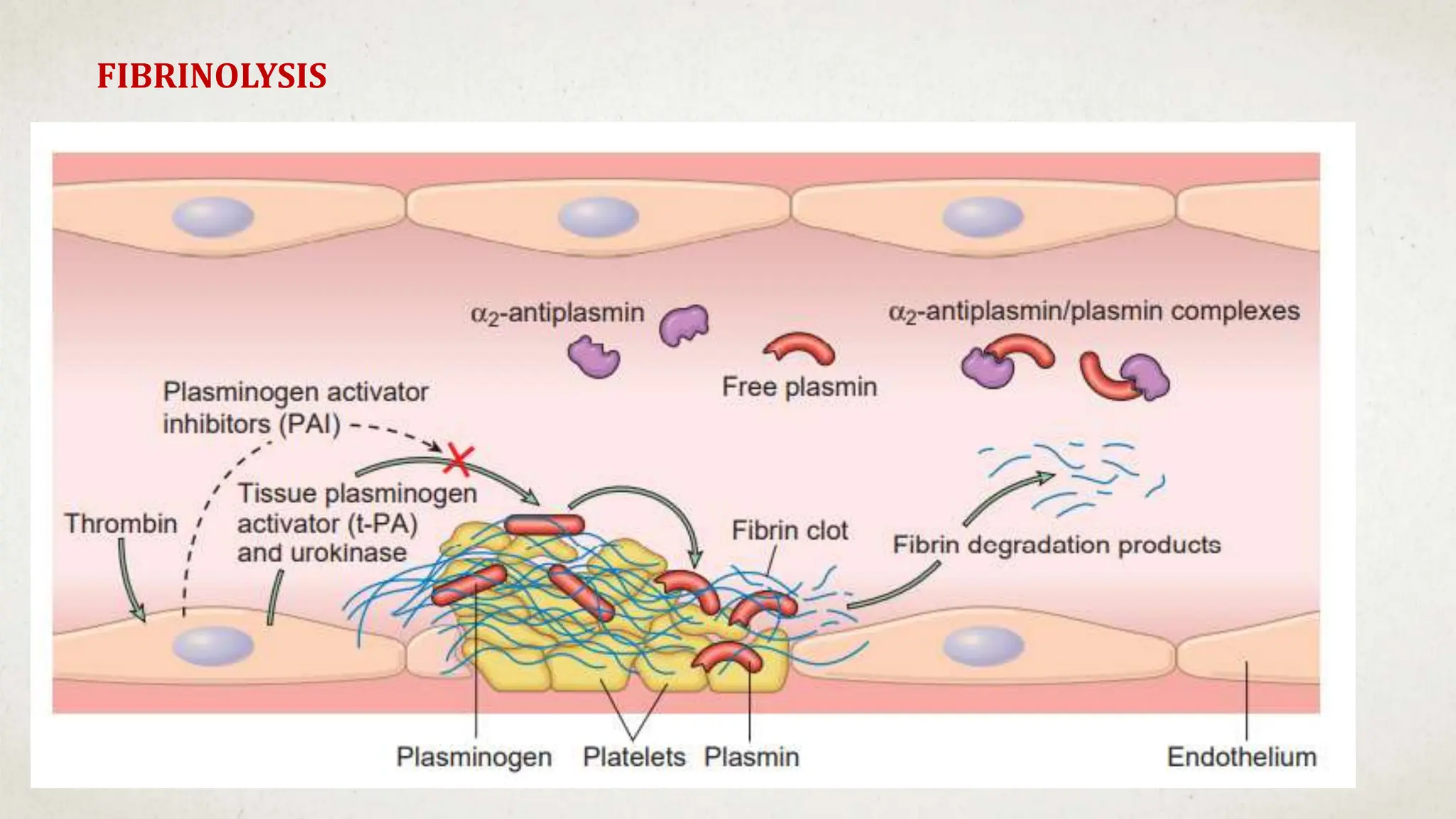

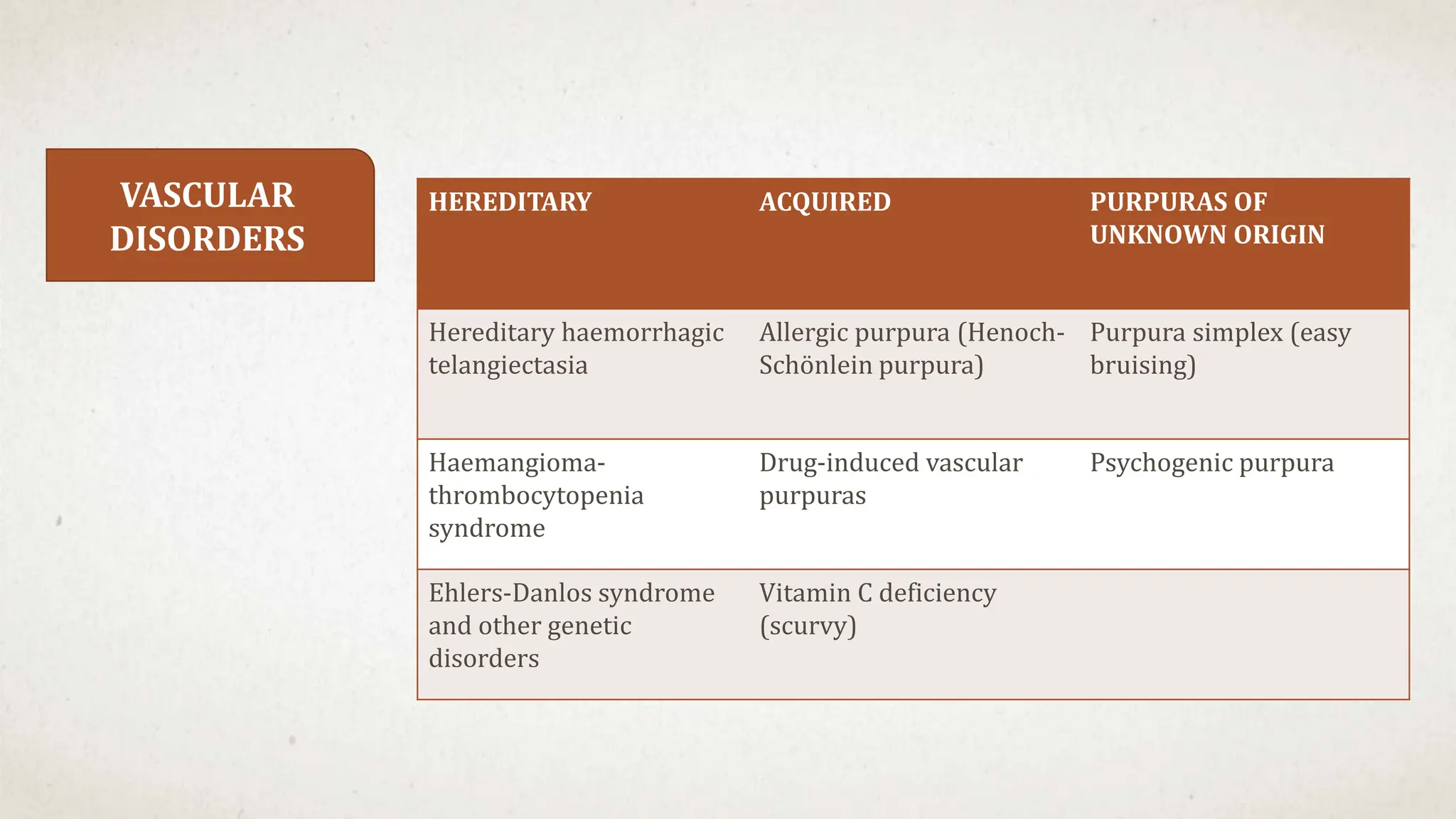

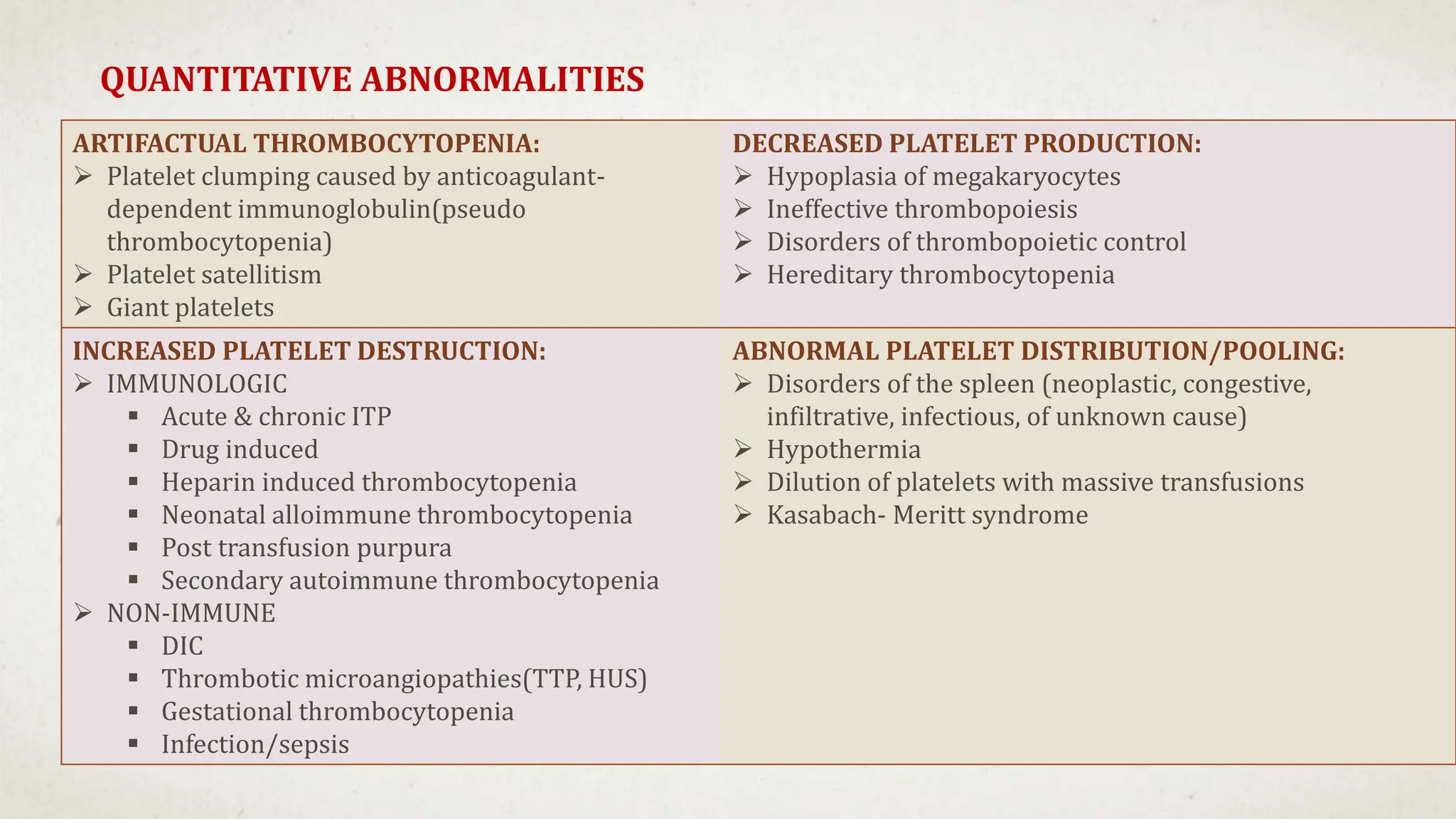





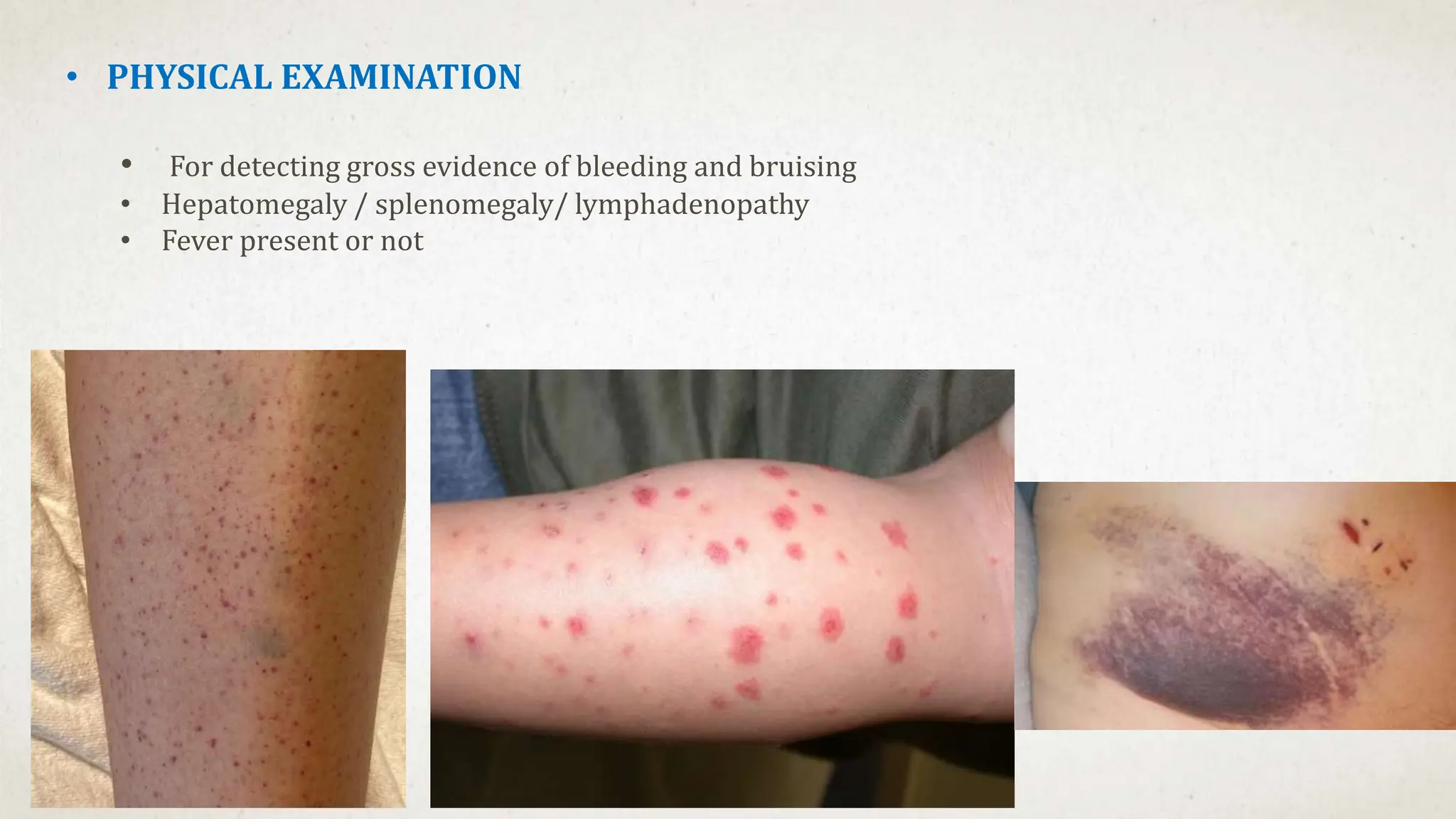
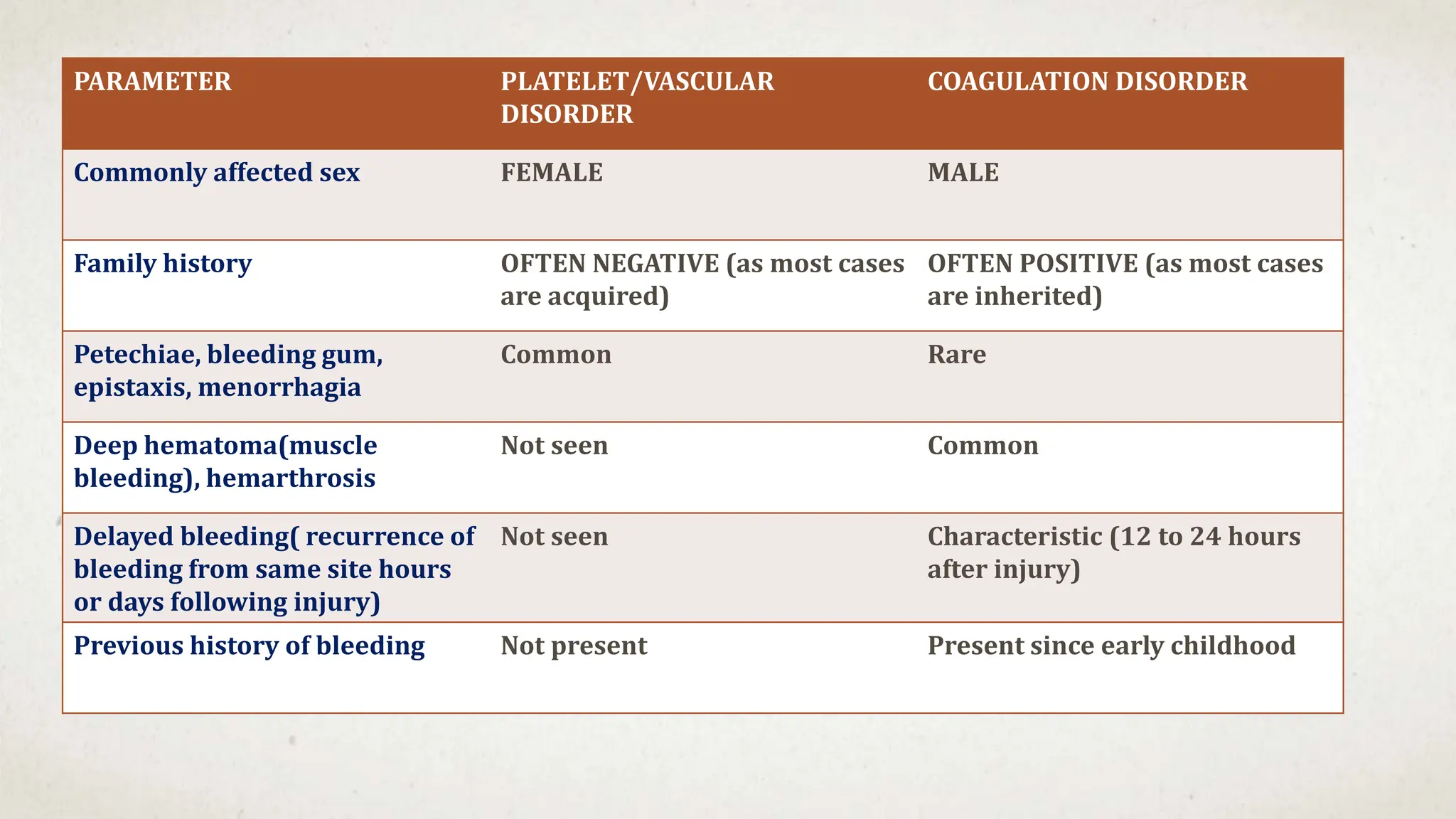

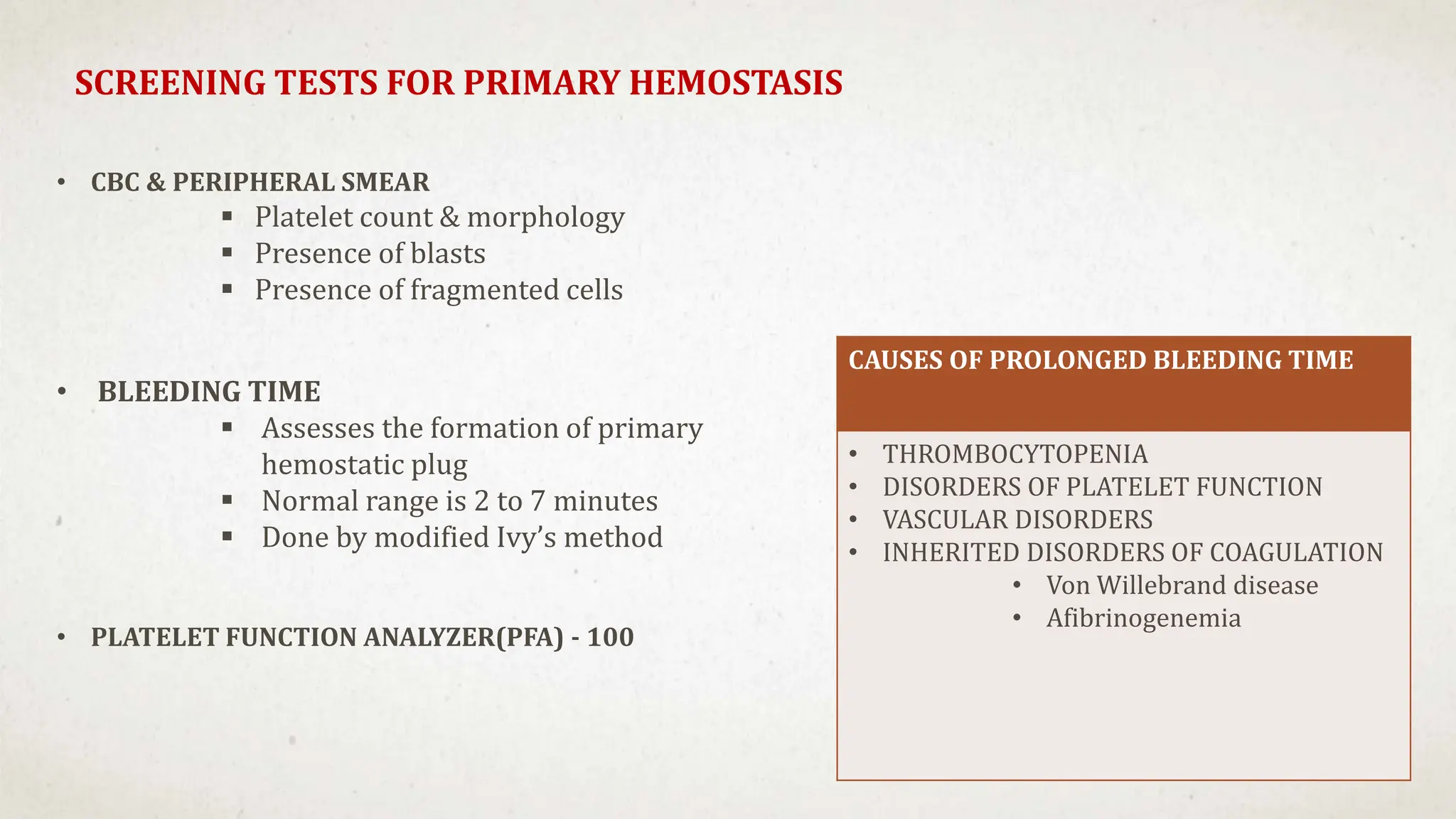

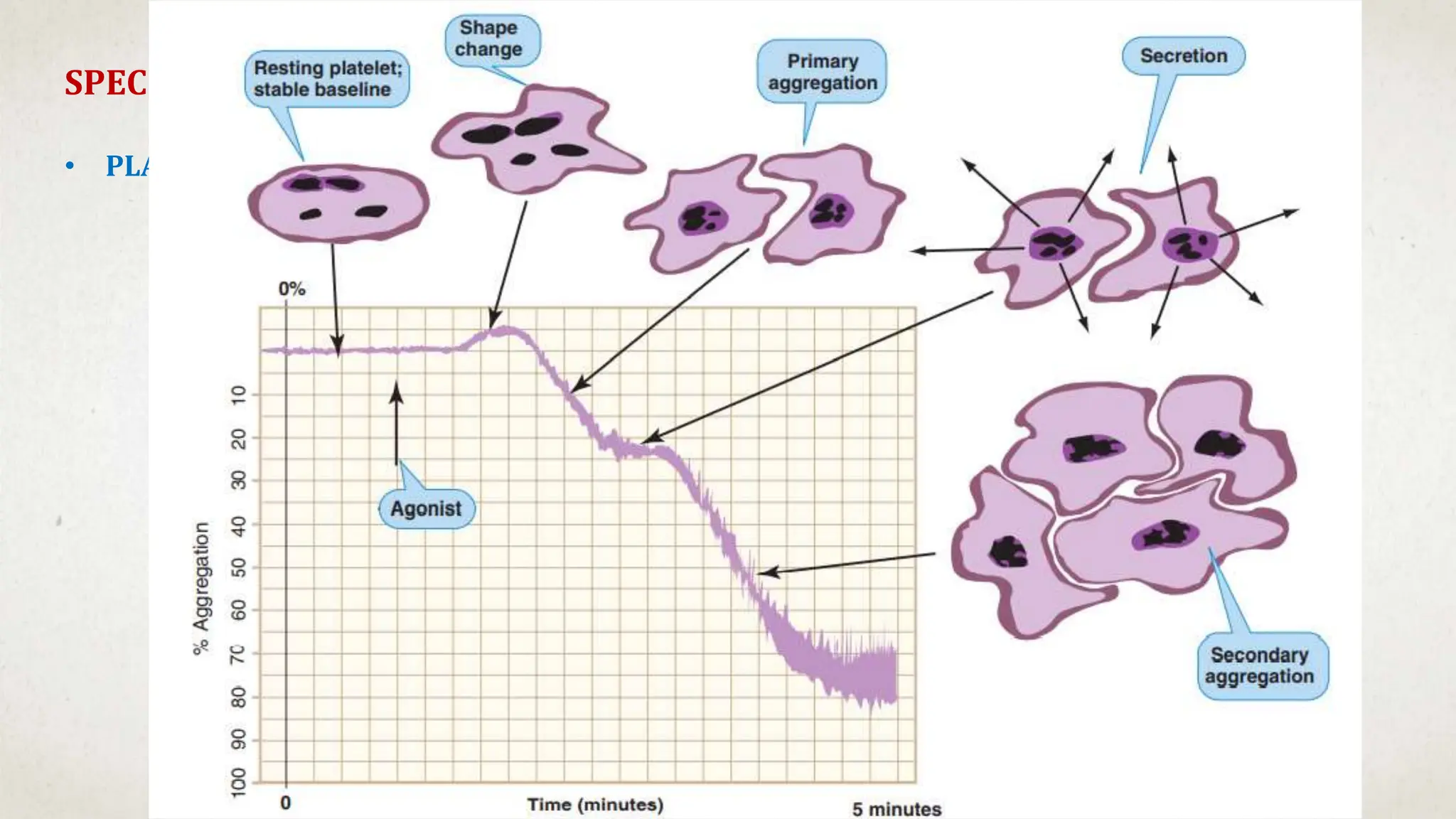






























































![Hematology%20disorder%20in%20dental%20treatment[1]](https://cdn.slidesharecdn.com/ss_thumbnails/hematology20disorder20in20dental20treatment1-161105033248-thumbnail.jpg?width=640&height=640&fit=bounds)





























