Downloaded 641 times





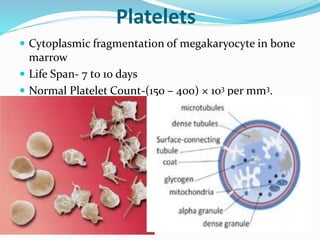
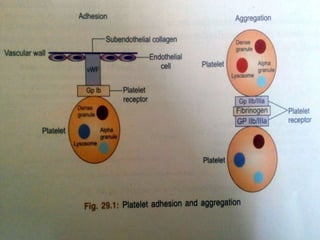


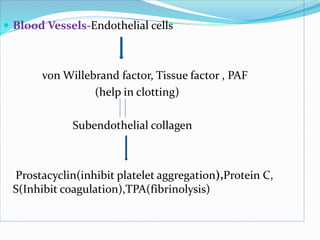

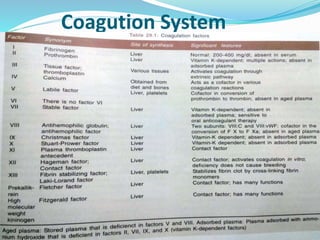
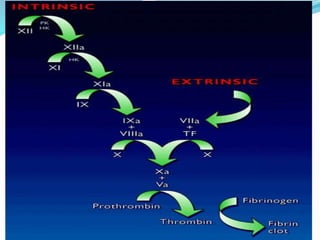
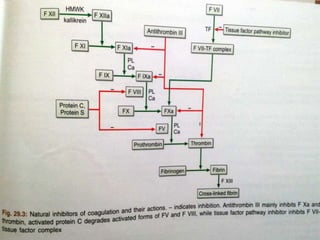

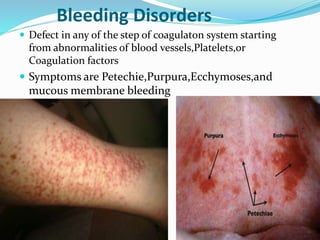



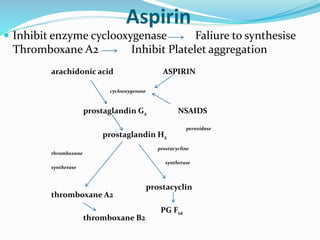








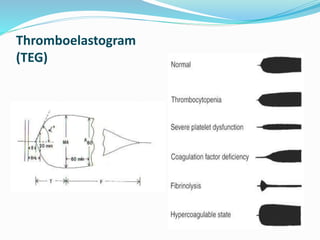


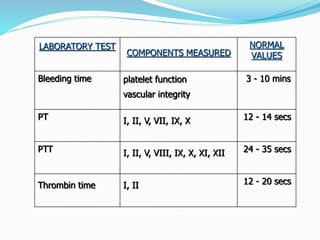


This document summarizes hemostasis and the coagulation process. It discusses that hemostasis involves platelets, blood vessels, and plasma proteins interacting to maintain blood fluidity and prevent hemorrhage. The three main steps of hemostasis are primary hemostasis involving platelet plug formation, the coagulation cascade, and fibrinolysis. Disorders can occur from abnormalities in blood vessels, platelets, or coagulation factors, causing excessive bleeding. Several laboratory tests are used to monitor coagulation factors and platelet function, including prothrombin time, activated partial thromboplastin time, and thromboelastography.



































































