Download as PDF, PPTX






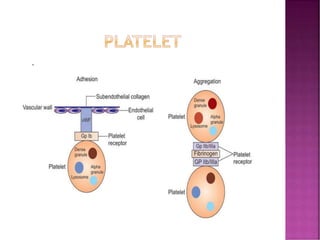




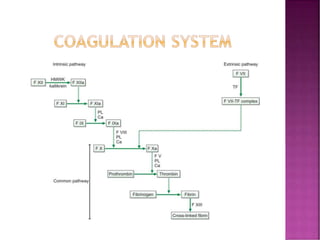
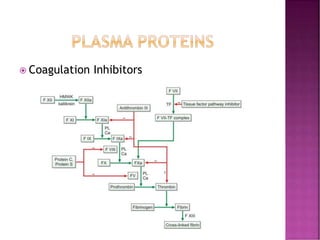
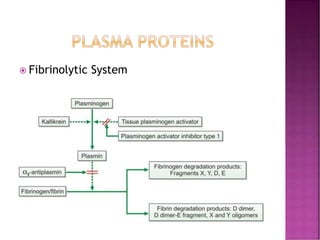





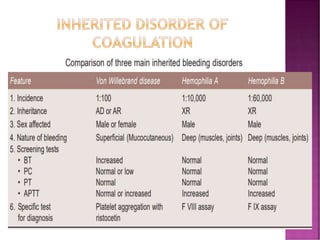
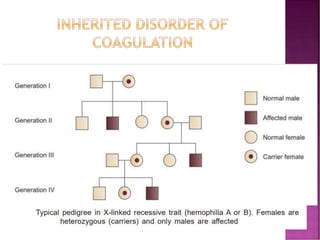





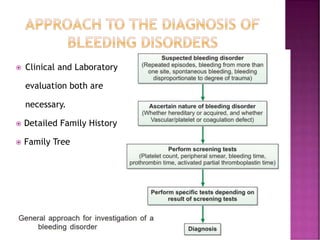
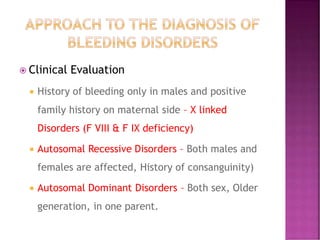


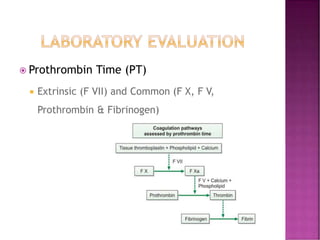




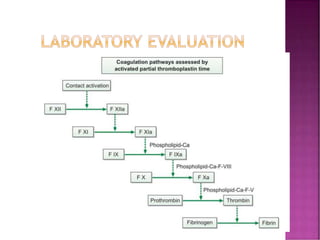


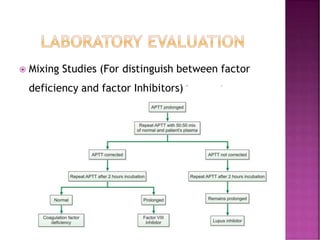







This document discusses bleeding disorders and their evaluation. It notes that bleeding disorders like hemophilia A and von Willebrand disease are common. A proper evaluation of a bleeding patient requires assessing whether the bleed is congenital or acquired, where the bleed is located, and other relevant clinical details. Laboratory tests that are part of the evaluation include complete blood count, prothrombin time, activated partial thromboplastin time, and specific coagulation factor assays. The document provides details on how these tests are performed and interpreted to diagnose specific coagulation factor deficiencies or other bleeding disorders.





















































