Download to read offline


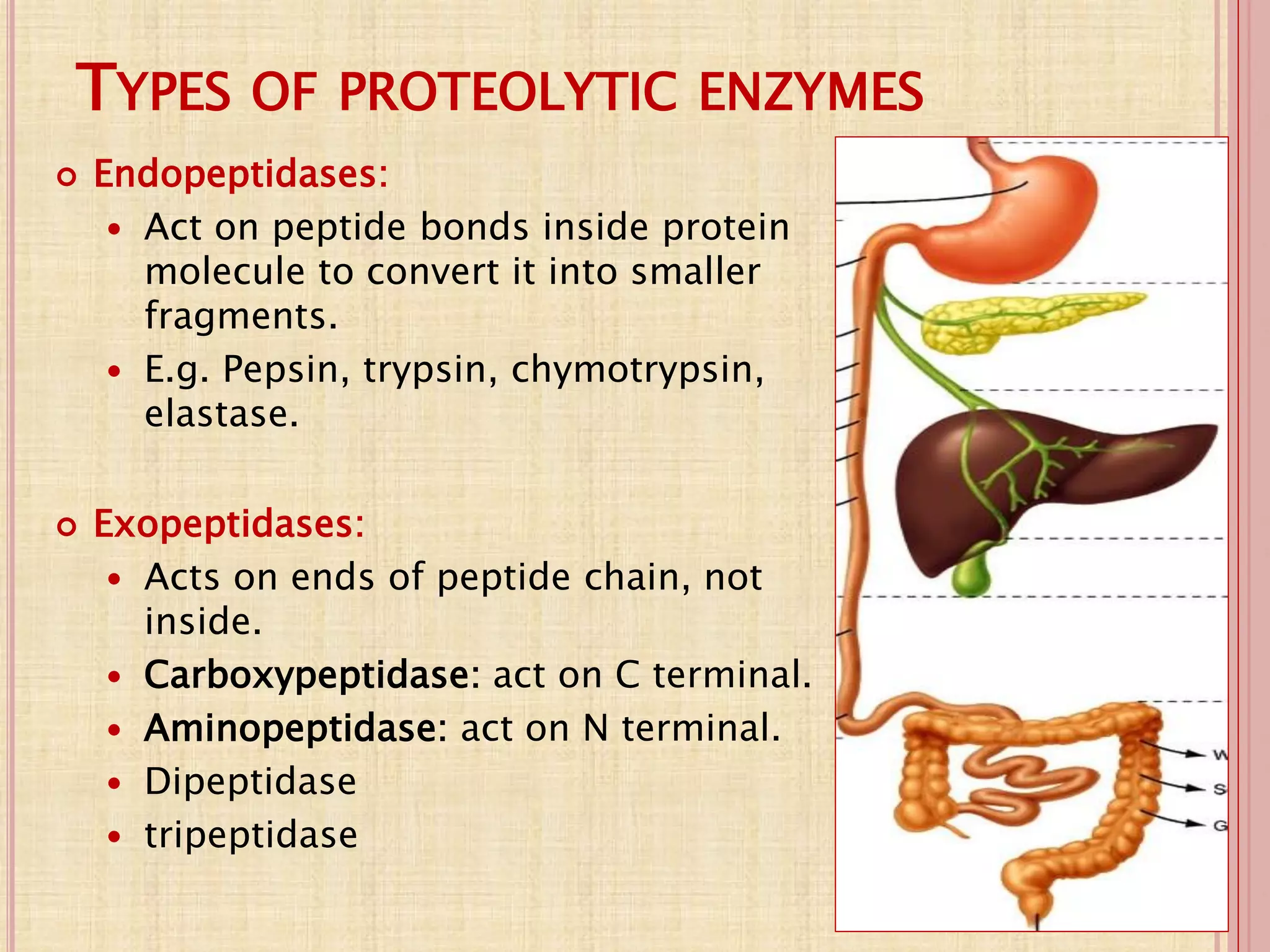









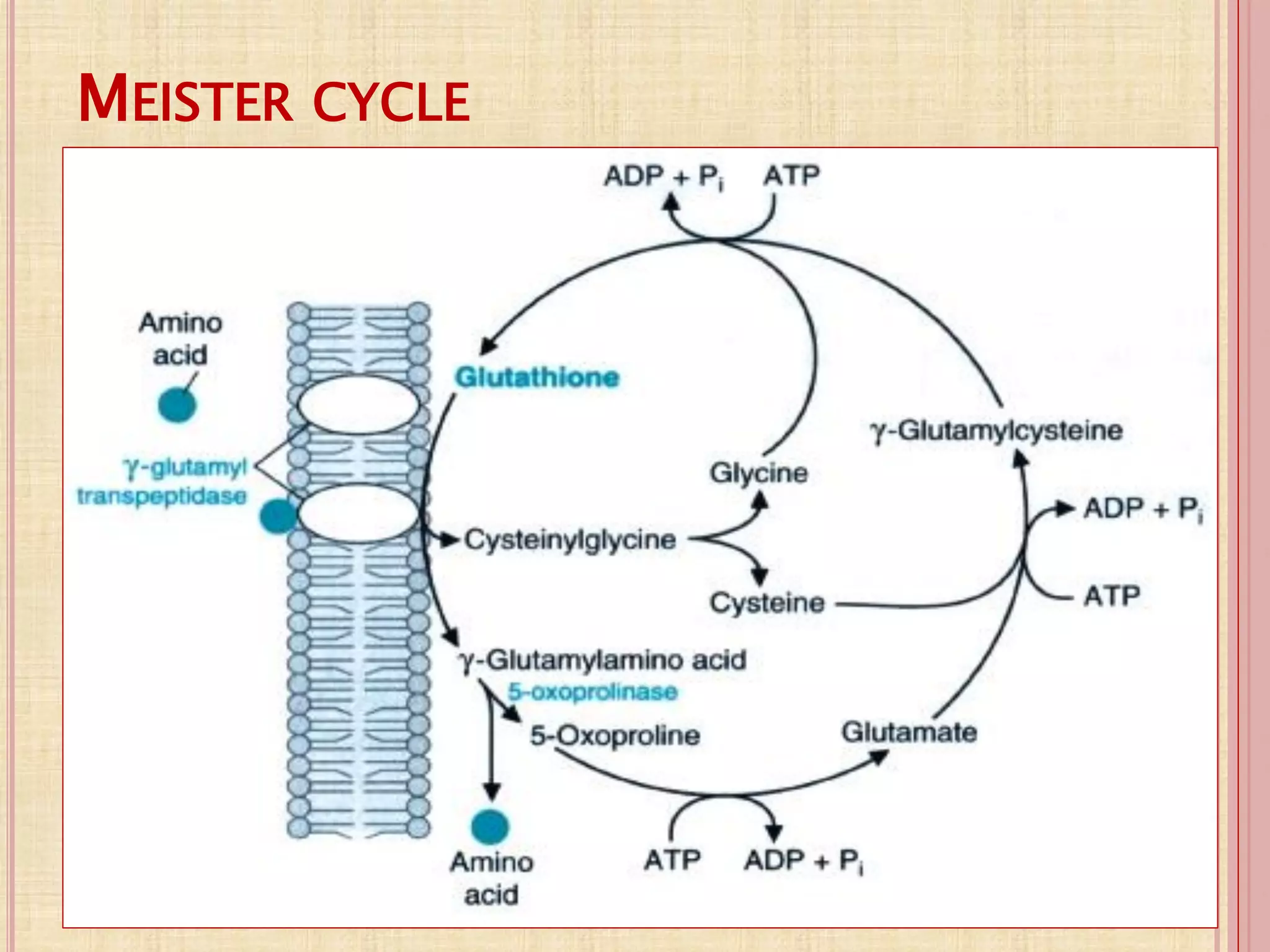




















(1) Protein digestion involves proteolytic enzymes that break proteins into smaller peptides and amino acids in the stomach and small intestine. (2) Amino acids are absorbed into the bloodstream and transported to tissues via three main mechanisms. (3) Within cells, amino acids undergo various reactions including transamination, oxidative and non-oxidative deamination, and transmethylation and trans-sulfuration. (4) Ammonia is a byproduct of amino acid metabolism and is shuttled to the liver where it is converted to urea to be excreted.



































































