Downloaded 121 times














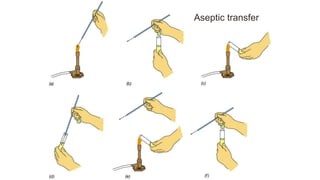
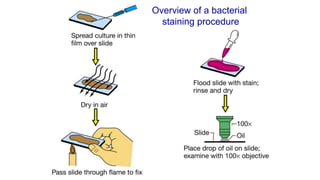
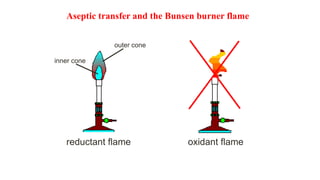












The document summarizes the Ziehl-Neelsen stain, an acid-fast stain used to identify acid-fast bacteria such as Mycobacteria. It works by using a primary stain, carbol fuchsin, which is retained by acid-fast bacteria after a decolorizing step. This allows acid-fast bacteria to be visualized as red rods against a blue counterstained background. The document discusses the staining procedure, interpretation of results, advantages and limitations of acid-fast staining, and causes of false-positive and false-negative results.























































































