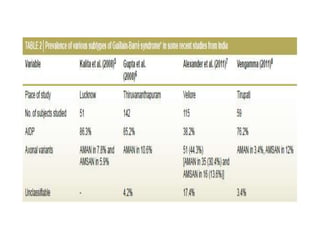
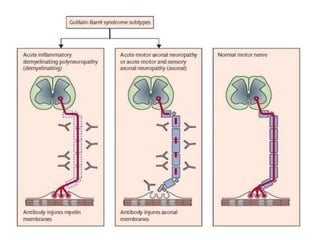
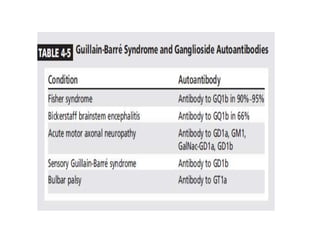
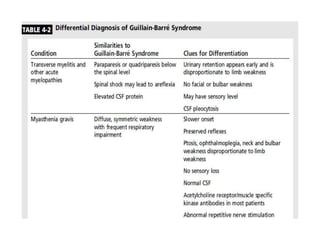
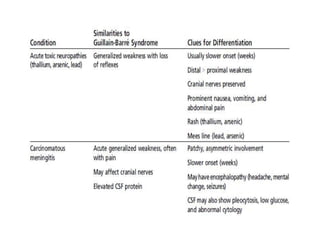
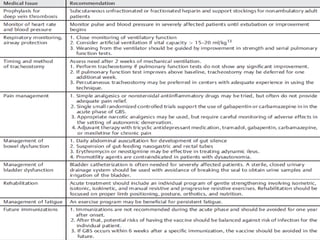
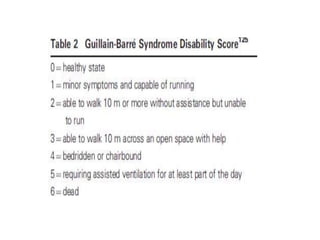
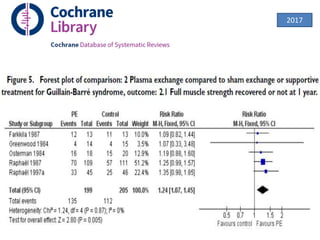
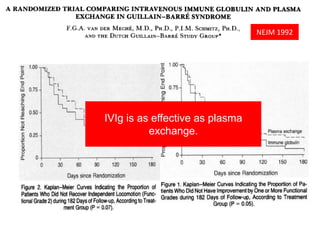
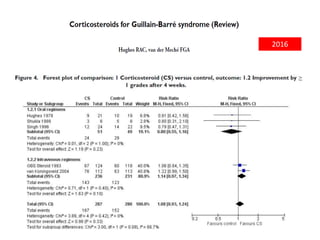
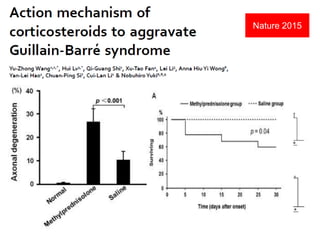
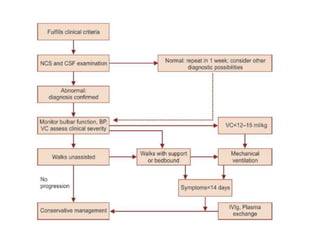
Dr. Nishtha Jain provides an overview of Acute Inflammatory Demyelinating Polyneuropathy (AIDP). Key points include: AIDP is an immune-mediated disorder of the peripheral nervous system, often preceded by a respiratory or gastrointestinal infection. Diagnosis involves lumbar puncture showing elevated CSF protein without pleocytosis. Electrodiagnosis can show features of demyelination. Treatment involves plasma exchange or IV immunoglobulin to remove antibodies. Prognosis is generally good, with most patients achieving near-full recovery, though respiratory failure can occasionally occur. New variants beyond classic AIDP have been recognized.












































![• One possible strategy for axon protection is sodium
channel blockade.
• Supporting this approach are data indicating flecainide
protects axons in an animal model (experimental
autoimmune neuritis [EAN]).
• Another general approach to axon protection, or
enhanced regeneration, is growth factor therapy.](https://image.slidesharecdn.com/aidp-171124154430/85/acute-inflammatory-demyelinating-polyneuropathy-56-320.jpg)






























































![DUAL AND TRIPLE ANTITHROMBOTIC THERAPY FOR SECONDARY STROKE [Autosaved].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/dualandtripleantithrombotictherapyforsecondarystrokeautosaved-230904113552-c3502b37-thumbnail.jpg?width=640&height=640&fit=bounds)






























![CASE_PRESENTATION_ON_subdural_hematoma(SDH)[1 FINAL PPT]-1.pptx](https://cdn.slidesharecdn.com/ss_thumbnails/casepresentationonsubduralhematomasdh1finalppt-1-260129172522-d405d375-thumbnail.jpg?width=640&height=640&fit=bounds)


