GUILLIAN BARRE SYNDROME
Guillain–Barré syndrome (GBS) is an acute onset, immune-mediated
disorder of the peripheral nervous system
GBS is now recognized as a heterogeneous syndrome with several
variant forms.
Most often, GBS presents as an acute, monophasic paralyzing illness
provoked by a preceding infection.
GBS is fairly rare. Statistics 1 to 3 per 100,000annually.
3.
HOW DOES ITPRESENT?
Progressive, symmetrical muscle weakness
Weakness starts in the legs, it begins in the
arms or facial muscles in about 10%
Weakness gradually worsens in ascending
fashion (Other variations exist)
Paresthesia in the hands and feet accompany
the weakness
Pain, typically located in the back and
extremities
Dysautonomia
4.
Initial symptoms Progressesover a period of about 2 weeks
Weakness may progress to complete paralysis of all extremity,
facial muscles
Respiratory and Bulbar muscle compromise
By 4 weeks 90% cases have reached the severity of the disease
Self limiting disease, and most patients recover.
Disease progression of more than 8 weeks is consistent with
Chronic Inflammatory Demyelinating Polyradiculoneuropathy
5.
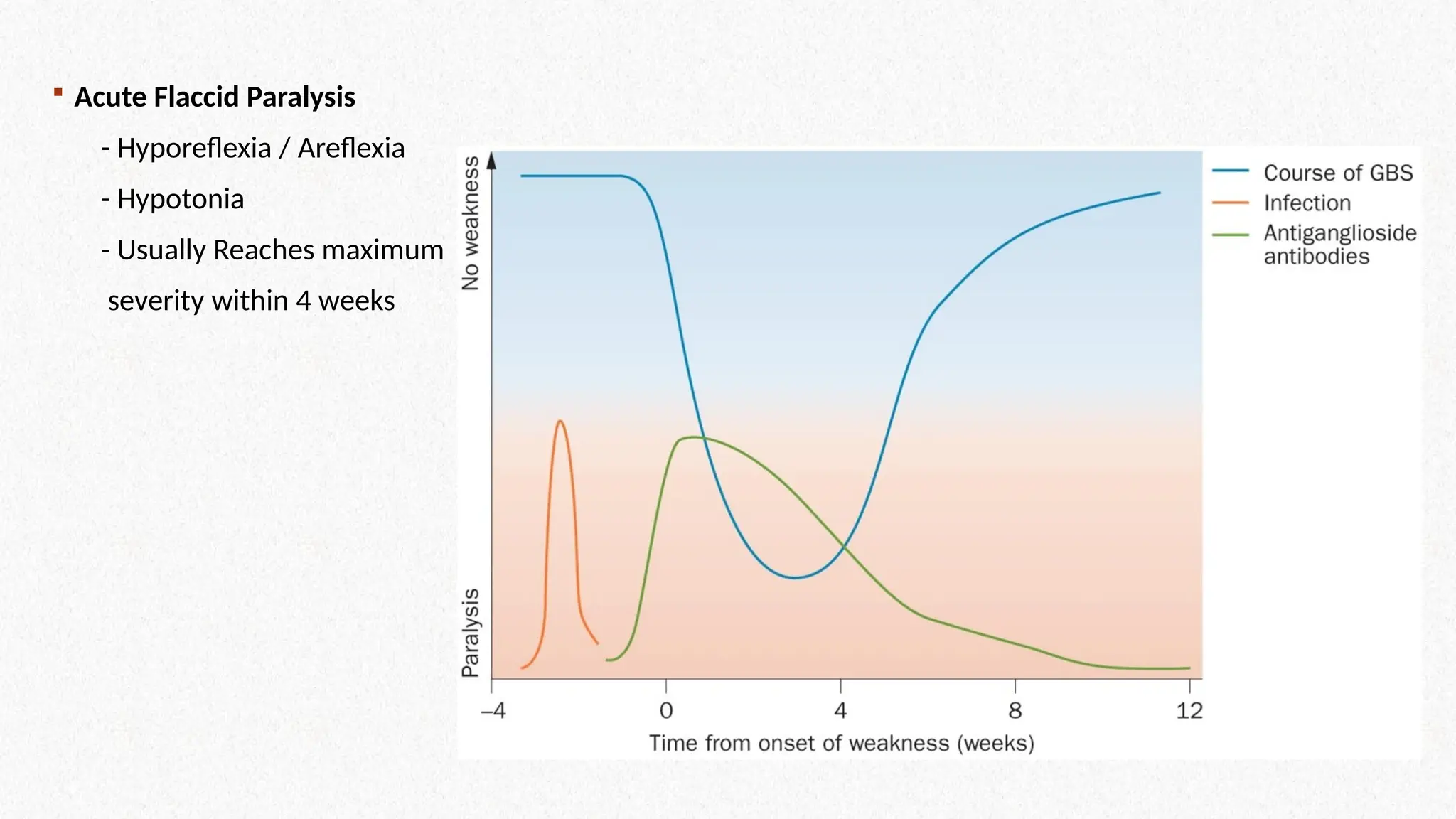
Acute FlaccidParalysis
- Hyporeflexia / Areflexia
- Hypotonia
- Usually Reaches maximum
severity within 4 weeks
6.
TYPES OF GBS
HistoricallyGBS was termed as one disease
More Subtypes / Variants discovered
Can be differentiated by nerve electrophysiological characteristics
and clinical presentation
1. Acute Inflammatory Demyelinating Polyradiculoneuropathy
(AIDP) (80-85%)
2. Acute Motor Axonal Neuropathy (AMAN)
3. Acute Motor and Sensory Axonal Neuropathy (AMSAN)
4. Miller Fisher Syndrome
Note: other rare variations are also documented, will not be focused on in this
CME
7.
RARE TYPES
Bickerstaff Encephalitis
PharyngealCervical Brachial Weakness
Acute pandysautonomia
Pure Sensory GBS
Facial Diplegia and Distal Limb Parasthesia
Acute Bulbar Palsy with Areflex
Note: These are rare types and will not be focused on in this CME
8.
HEALTH OUTCOMES
In TreatedCase
Respiratory failure requiring ventilation in about 25% Patients
Death in 4 – 15% of GBS Patients
Persistent Disability in about 20% patients with GBS
Persistent Fatigue in 67% Patients
9.
GUILLIAN BARRE SYNDROMESYBTYPES
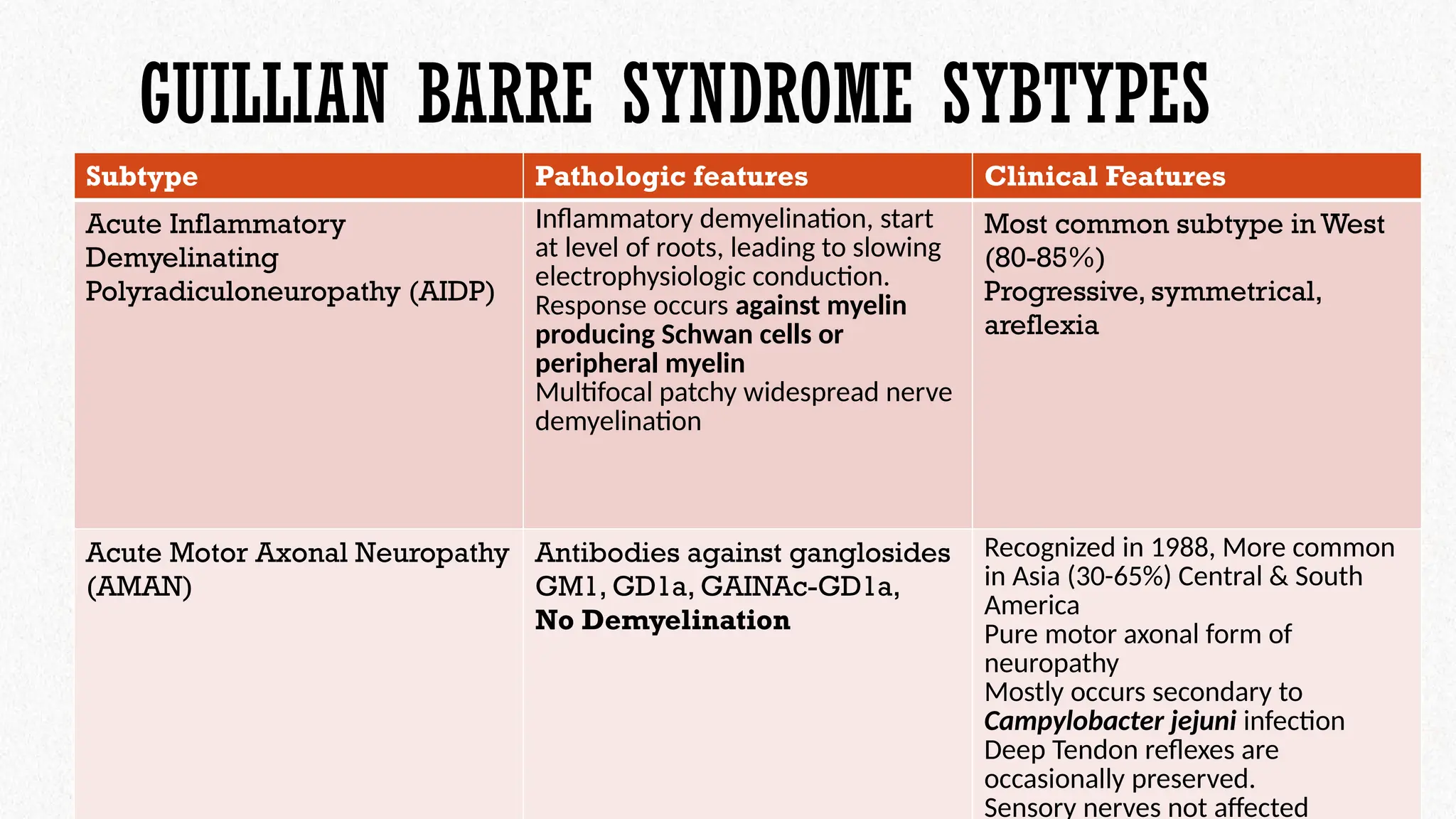
Subtype Pathologic features Clinical Features
Acute Inflammatory
Demyelinating
Polyradiculoneuropathy (AIDP)
Inflammatory demyelination, start
at level of roots, leading to slowing
electrophysiologic conduction.
Response occurs against myelin
producing Schwan cells or
peripheral myelin
Multifocal patchy widespread nerve
demyelination
Most common subtype in West
(80-85%)
Progressive, symmetrical,
areflexia
Acute Motor Axonal Neuropathy
(AMAN)
Antibodies against ganglosides
GM1, GD1a, GAINAc-GD1a,
No Demyelination
Recognized in 1988, More common
in Asia (30-65%) Central & South
America
Pure motor axonal form of
neuropathy
Mostly occurs secondary to
Campylobacter jejuni infection
Deep Tendon reflexes are
occasionally preserved.
Sensory nerves not affected
10.
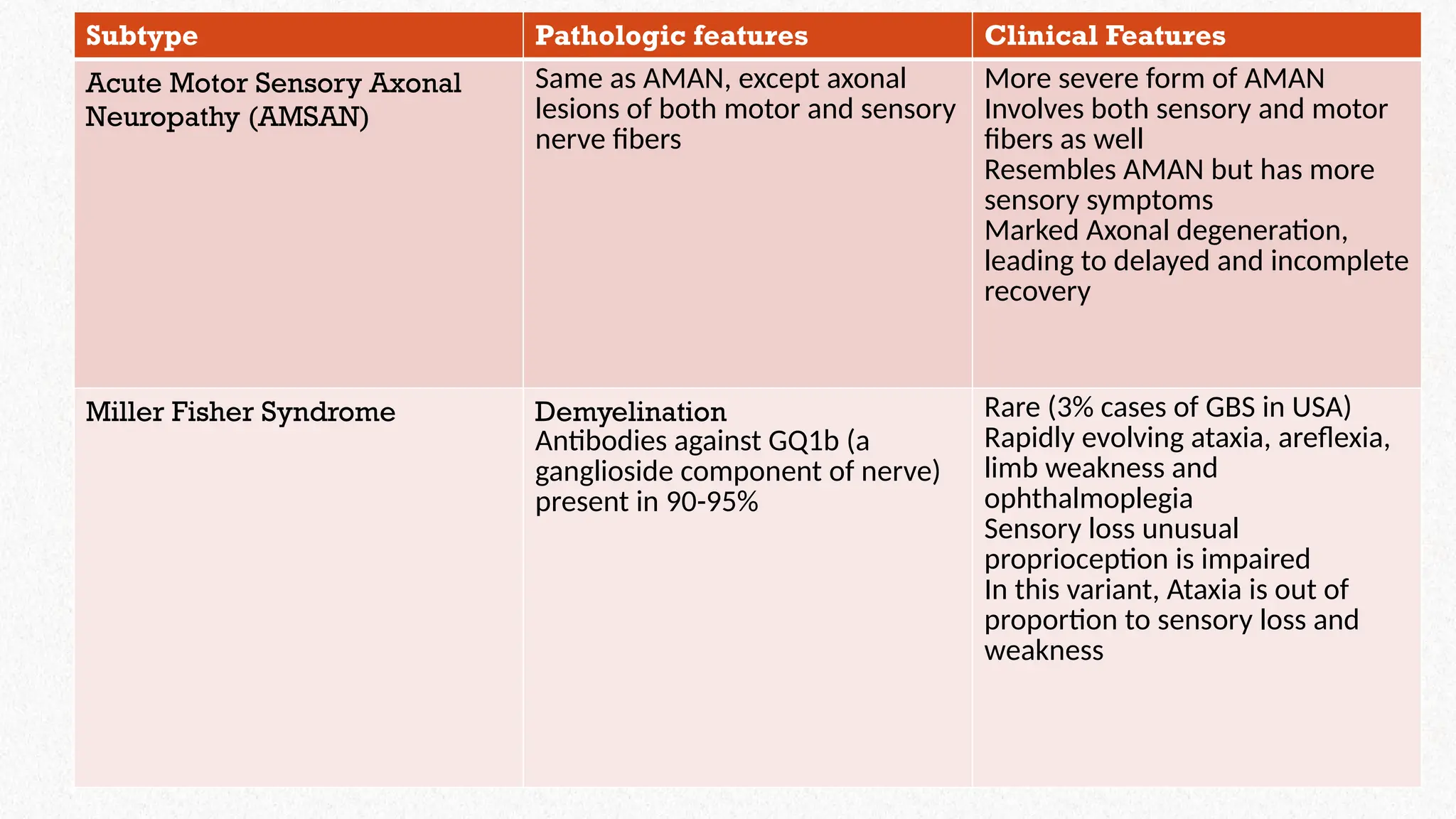
Subtype Pathologic featuresClinical Features
Acute Motor Sensory Axonal
Neuropathy (AMSAN)
Same as AMAN, except axonal
lesions of both motor and sensory
nerve fibers
More severe form of AMAN
Involves both sensory and motor
fibers as well
Resembles AMAN but has more
sensory symptoms
Marked Axonal degeneration,
leading to delayed and incomplete
recovery
Miller Fisher Syndrome Demyelination
Antibodies against GQ1b (a
ganglioside component of nerve)
present in 90-95%
Rare (3% cases of GBS in USA)
Rapidly evolving ataxia, areflexia,
limb weakness and
ophthalmoplegia
Sensory loss unusual
proprioception is impaired
In this variant, Ataxia is out of
proportion to sensory loss and
weakness
11.
WHAT TRIGGERS THISAUTOIMMUNE RESPONSE?
Bacterial
1. Campylobacter Jejuni
2. Mycoplasma Pneumonia
3. H.Influenzae
4. Borealla
5. Brucella
Viral
Cytomegalovirus
Herpes Simplex
Varicella Zoster
Epstein Barr
Hep A,B,C,E
HIV Virus
Zika Virus
** Flu Vaccine – (Very rare. Cases only reported in 1-2 per million vaccination)
12.
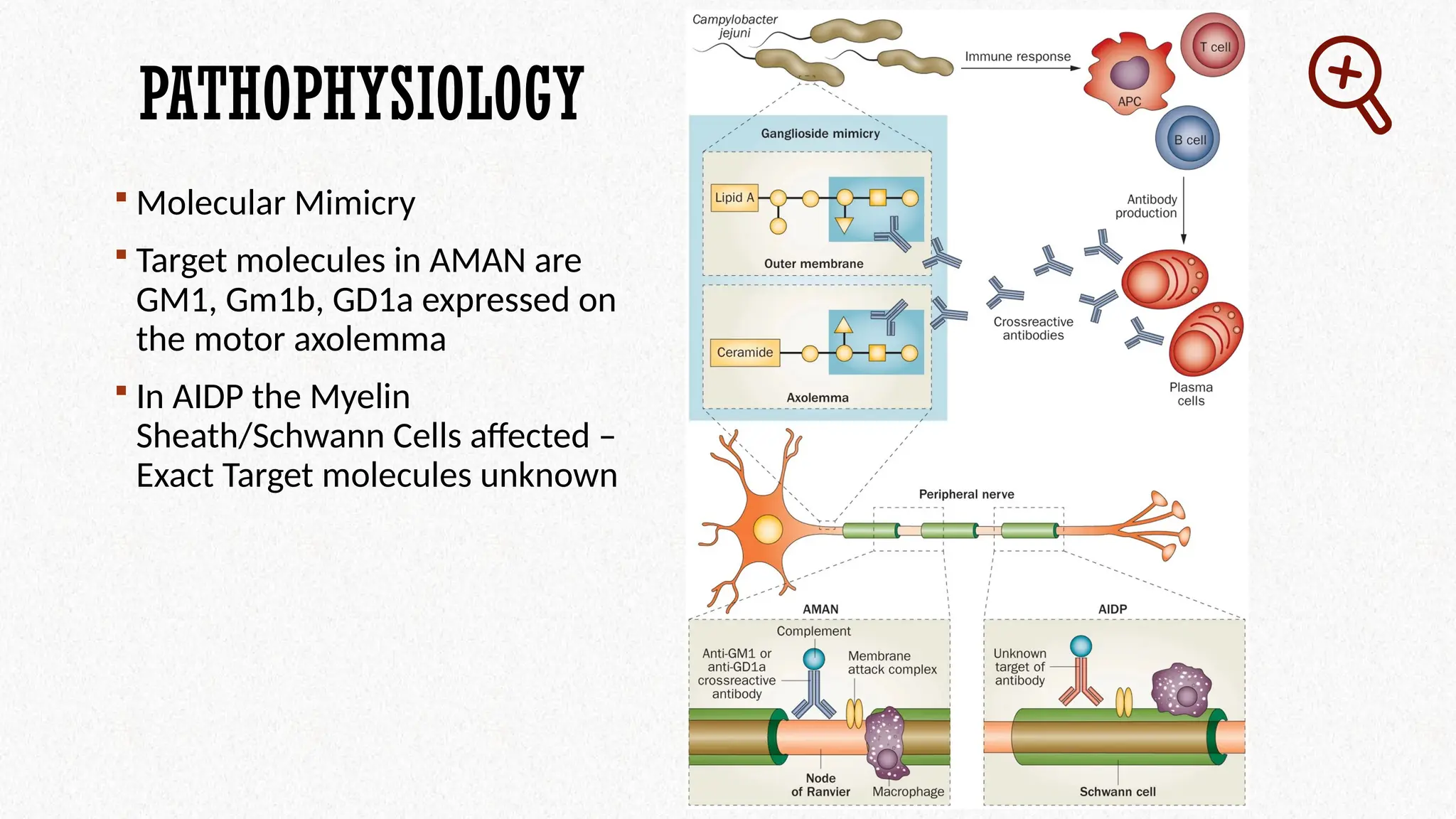
PATHOPHYSIOLOGY
Molecular Mimicry
Target molecules in AMAN are
GM1, Gm1b, GD1a expressed on
the motor axolemma
In AIDP the Myelin
Sheath/Schwann Cells affected –
Exact Target molecules unknown
BASIC OF NCV
Definitionof Latency:
Latency is the time (in milliseconds) between:
The electrical stimulus applied to a nerve and The initial response recorded by
the electrode (either over a muscle or sensory nerve)
15.
NCV FINDINGS INGBS
Slow CT is the mnemonics used to remember the NCV findings
16.
DIAGNOSTIC APPROACH
Historyand Clinical Presentation
Nerve Conduction Studies (NCS) + Needle
electromyography (EMG)
May be normal in up to 13 % soon after
symptom onset,
but rarely remain normal on sequential
testing over the initial weeks of
symptoms.
Demyelinating forms of GBS – Decreased
motor nerve conduction velocity,
prolonged distal motor latency, Increased
F wave latency, conduction Blocks
Lumbar Puncture
Elevated CSF Protein with a normal CSF
white cell count
(Alubminocytologic
Dyssociation)
A normal CSF protein is found in 1/3 to
1/2 patients when tested within one week
But they are elevated in 90% of patients
by the end of the second week of
symptoms
17.
DIAGNOSTIC APPROACH
Antibodies
MRI
Serum IgG antibodies for GQ1b is useful for diagnosis of Miller Fisher Sydrome
Spinal MRI may reveal thickening and enhancement of intrathecal spinal roots and
cauda equina.
Also helpful in ruling out GBS or other cause? focal spinal lesion or other injury
could indicate other source of weakness i.e Transverse Myelitis
18.
INITIAL ASSESSMENT +COMPLICATIONS
Assess degree of weakness, proper history and recent illnesses / trauma, Spinal Cord Injury, day of weakness
Check for pattern of weakness and symmetry
Neuromuscular Respiratory failure – May need Mechanical Ventilation
Criteria for Elective Intubation
Forced Vital Capacity <20ml/kg
Maximum Inspiratory Pressure <30 cmH20
Maximum Expiratory Pressure <40 cmH20
Predictors for Respiratory Failure
Inability to cough, stand, lift head
Deranged LFTs
Presence of Facial or Bulbar weakness
19.
MONITORING OF FVC
Forced Vital Capacity and Maximum Inspiratory Pressure
Frequent evaluations of these parameters should be performed at bedside – to
assess need for ventilatory assistance – Can be done daily
Forced Vital Capacity is very helpful as can determine when to Intubate
Maximum Inspiratory Pressure / Negative Inspiratory Force (NIF) - bedside test
can be done every half-hourly.
(Normal is greater than 60 cm Water)
20.
TREATMENT
Two therapyOptions are available
1) IVIG Administration
Exact Mechanism unknown. Theorized to interfere with activation of
complement and production of cytokines and interfering with activation
and effector function of T and B Cells
Given 0.4g/kg per day for consecutive 5 days.
2 g/kg total, given over 5 days
That is 0.4 g/kg/day × 5 days
2) Plasma Exchange / Plasmapheresis
Removes circulating antibodies, complement and biological response
modifiers.
Most effective when started early
21.
WHICH IS BETTER?IVIG VS PE
American Academy of Neurology (AAN) Recommendations for Immunotherapy/IVIG
PLASMAPHERESIS
Author /
Year
Class Results
French
Co-op
Group on
plasma
exchange
in GBS,
1997
Compare PE with
supportive
treatment
II RCT In the PE group, time to onset
of motor recovery was
significantly shortened
compared to the control group
(p=0.0002).
The number of patients with
one or more grades of
improvement at one month
22.
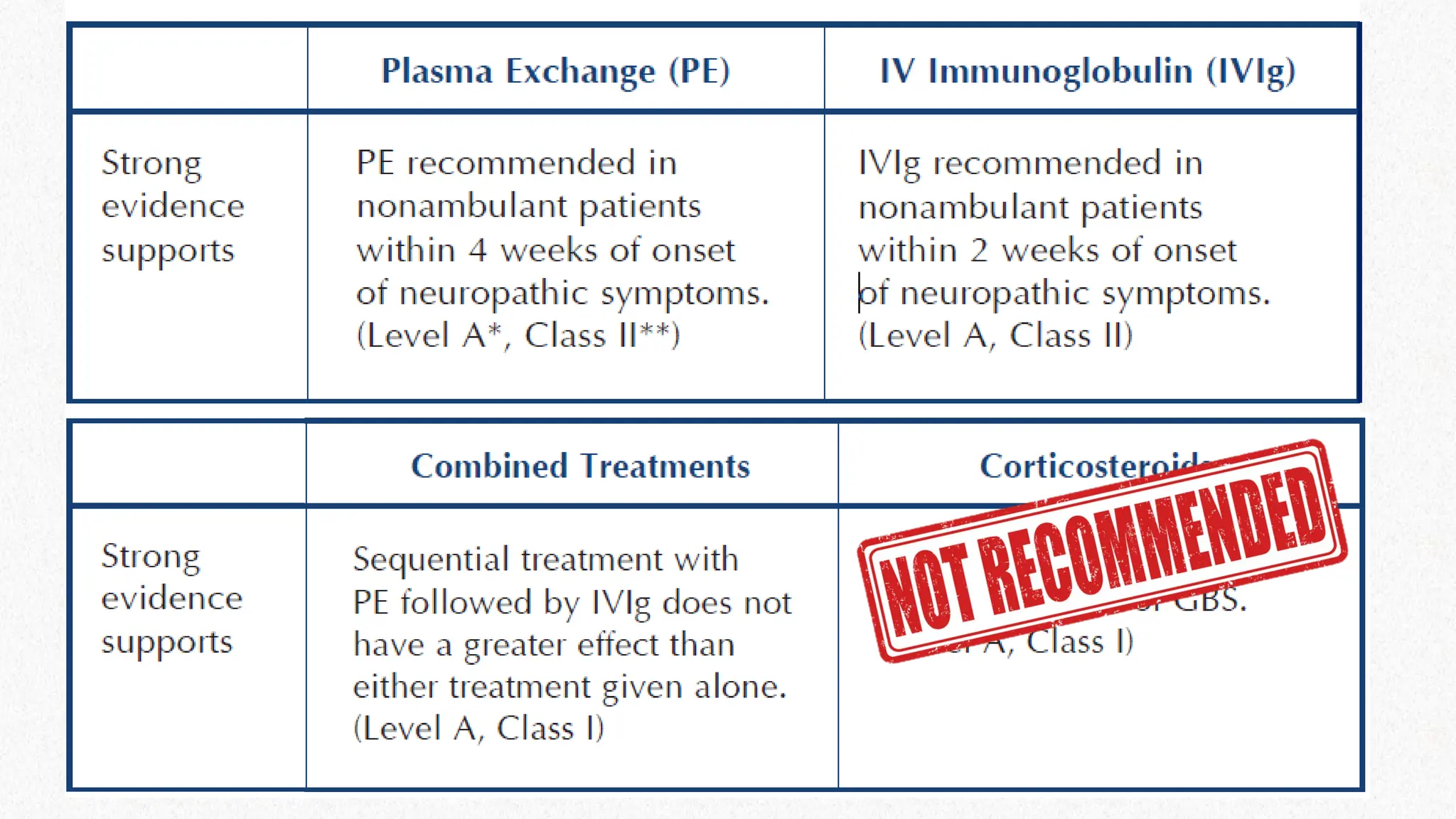
Plasma exchange hastensrecovery in non-ambulant patients
Plasa exchange also hastens recovery in ambulant patients within 2
weeks (limited to 1 study)
Plasmapheresis is recommended in non-ambulant patients within 4
weeks from onset
Recommended for ambulant patients within 2 weeks from onset
23.
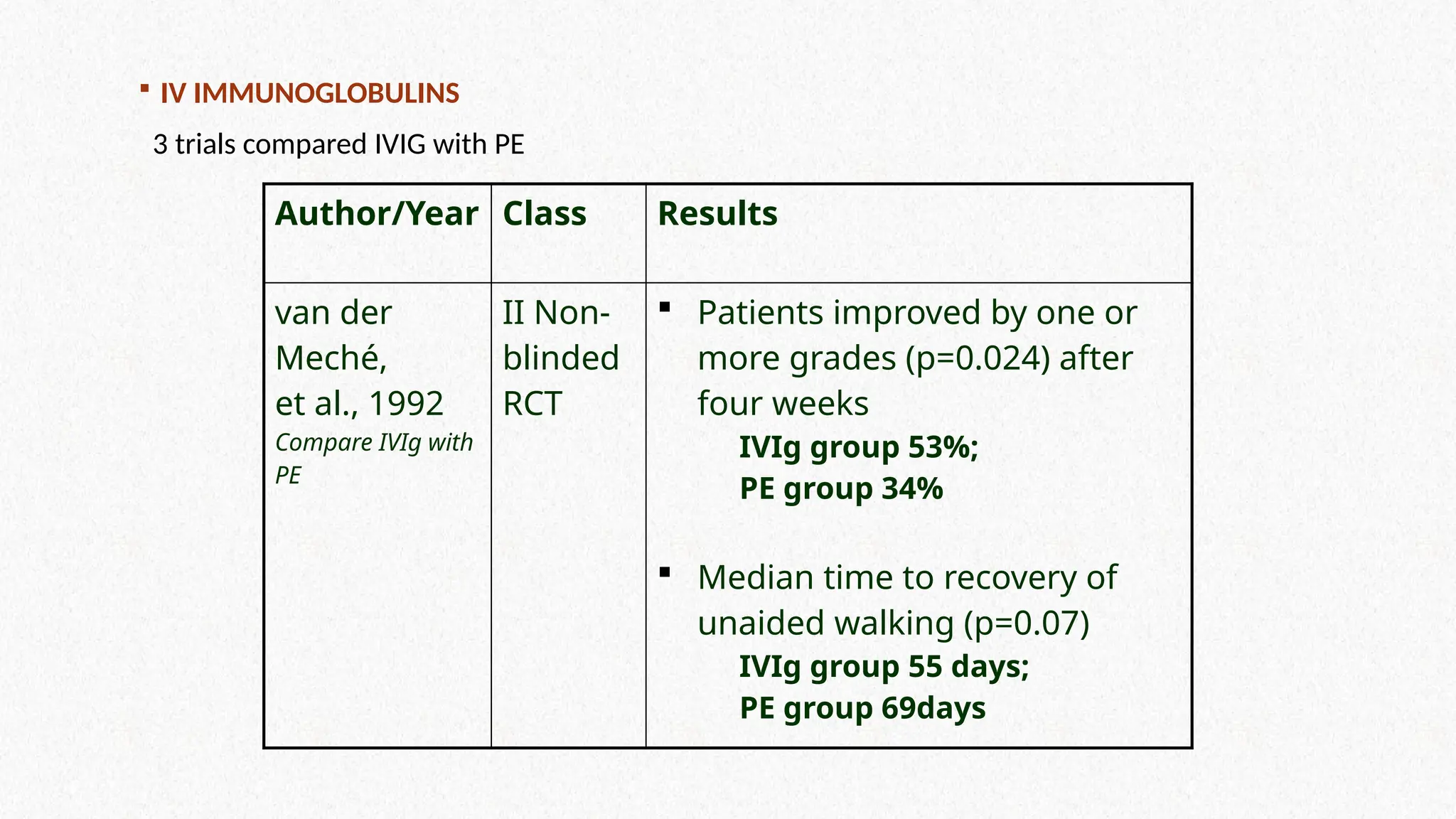
IV IMMUNOGLOBULINS
3trials compared IVIG with PE
Author/Year Class Results
van der
Meché,
et al., 1992
Compare IVIg with
PE
II Non-
blinded
RCT
Patients improved by one or
more grades (p=0.024) after
four weeks
IVIg group 53%;
PE group 34%
Median time to recovery of
unaided walking (p=0.07)
IVIg group 55 days;
PE group 69days
24.
Author/Year Class Results
Bril,et al., 1996
Compare IVIG with
supportive treatment
II RCT Median time to recover
ability to do manual work
IVIg group 65 days;
PE group 90 days
Mean disability grade
improvement
IVIg group 1.2;
PE group 1.0
IV IMMUNOGLOBULINS
Trial Compared with Supportive Treatment
25.
CONCLUSION
IVIG has equivalentefficacy to PE in hastening recovery from
patients requiring aid
Multiple complications less frequent than PE
IVIG is recommended for patients who require aid to walk within 2
– 4 weeks from onset of symptoms
The effects of IVIG and PE are equivalent.
One Class trial – PE followed by IVIG – No Significant Benefit
26.
CORTICOSTEROIDS?
Multiple studiesdone, including Intramuscular ACTH, IV Methylprednisolone, Oral
Prednisolone or Predinsone
Author/Year Class Results
Mendell et
al., 1985
Effect of plasma
exchange and
prednisone
II Alternate
allocation
controlled
trial
No significant difference in
any outcome
Shukla et al.,
1988
Effect of
prednisolone
I RCT No significant difference in
any outcome
27.
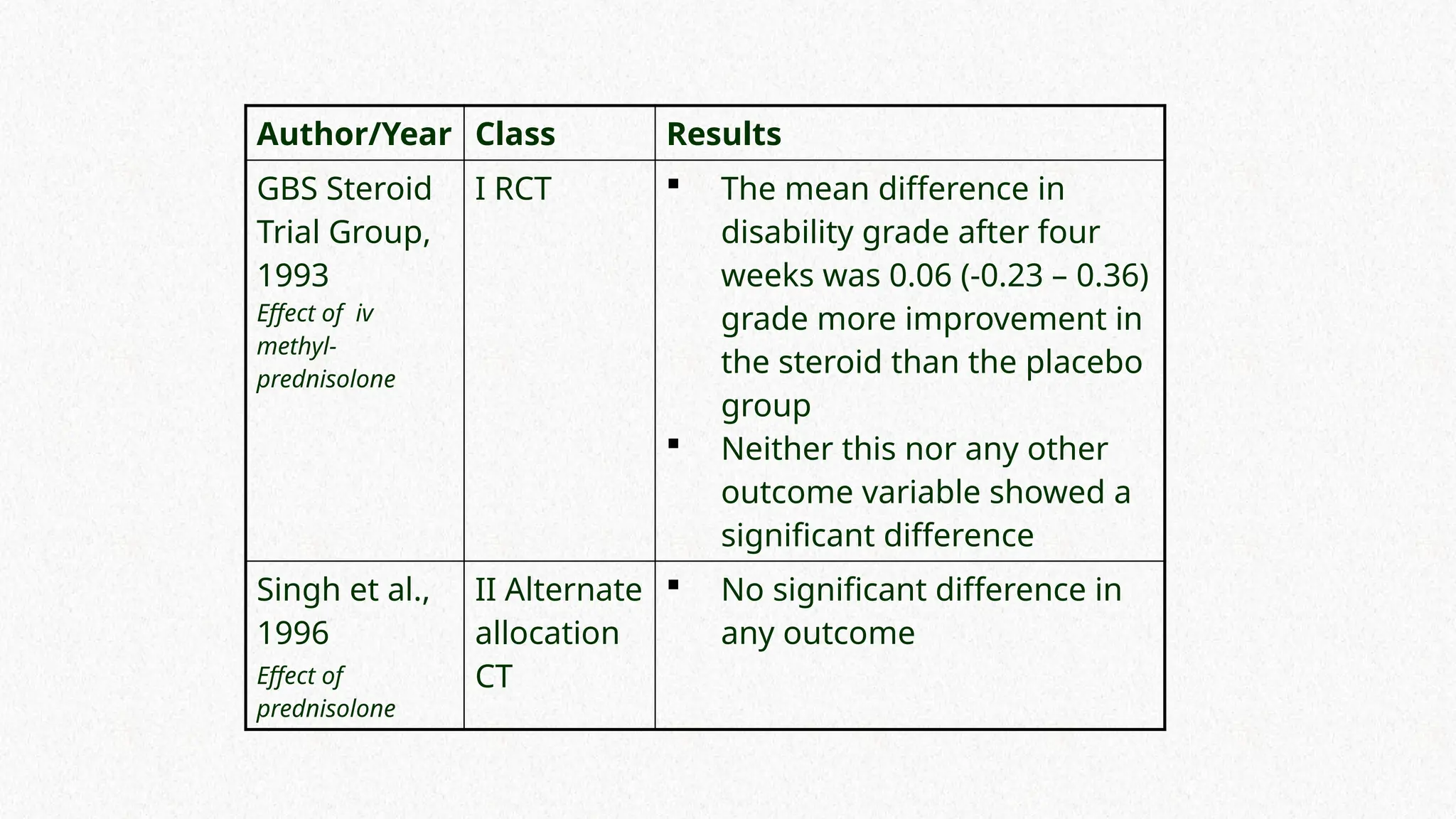
Author/Year Class Results
GBSSteroid
Trial Group,
1993
Effect of iv
methyl-
prednisolone
I RCT The mean difference in
disability grade after four
weeks was 0.06 (-0.23 – 0.36)
grade more improvement in
the steroid than the placebo
group
Neither this nor any other
outcome variable showed a
significant difference
Singh et al.,
1996
Effect of
prednisolone
II Alternate
allocation
CT
No significant difference in
any outcome
29.
MANAGEMENT / TREATMENT
SupportiveCare
Close Monitoring / ICU Setting in initial stage
Intubation in case of Respiratory Failure
Cardiovascular Management – Autonomic Dysfunction
DVT Prophylaxis, Bladder, Bowel Care, Psychological support,
Physiotherapy
Quadriplegic patients – Pressure Ulcer Prophylaxis
Pain Management
Rehabilitation
30.
PROGNOSTIC FACTORS
Older Age
RapidOnset (less than 7 days)
Severe muscle weakness
Need for ventilatory support
Average distal motor response amplitude reduction to <20%
normal
Preceding diarrheal illness
31.
RELAPSES
About 2-5% ofpatients will develop chronic relapsing weakness of
CIDP
Might be difficult to distinguish between GBS releated fluctuation
and acute onset CIDP
Points in favor of CIDP Rather than GBS:
Deterioration after 8 weeks from onset of weakness
Deterioration >3 times
No loss of independent ambulation
Absence of cranial nerve involvement
32.
REFERENCES
UptoDate
Medscape
American Academy of Neurology (AAN) guideline on GBS
www.aafp.org/ffp
https://doi.org/10.1002/brb3.960