Downloaded 22 times

























































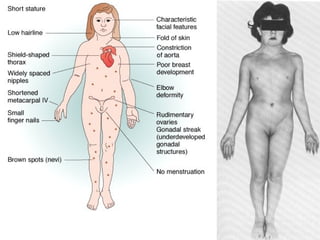




This document summarizes genetic disorders and mutations. It discusses single gene mutations that follow Mendelian inheritance patterns, including autosomal dominant, autosomal recessive, and sex-linked disorders. It also describes chromosomal disorders like trisomy 21 (Down syndrome) and Klinefelter syndrome. Additionally, it covers multifactorial inheritance and provides examples of genetic disorders caused by mutations, deletions, expansions, and other changes to DNA.



















































































