Downloaded 45 times





![Control Structures
if condition:
statements
[elif condition:
statements] ...
else:
statements
while condition:
statements
for var in sequence:
statements
break
continue](https://image.slidesharecdn.com/2015bioinformaticsbiopythonpart3-151116134050-lva1-app6892/85/2015-bioinformatics-bio_python_part3-6-320.jpg)
![Lists
• Flexible arrays, not Lisp-like linked
lists
• a = [99, "bottles of beer", ["on", "the",
"wall"]]
• Same operators as for strings
• a+b, a*3, a[0], a[-1], a[1:], len(a)
• Item and slice assignment
• a[0] = 98
• a[1:2] = ["bottles", "of", "beer"]
-> [98, "bottles", "of", "beer", ["on", "the", "wall"]]
• del a[-1] # -> [98, "bottles", "of", "beer"]](https://image.slidesharecdn.com/2015bioinformaticsbiopythonpart3-151116134050-lva1-app6892/85/2015-bioinformatics-bio_python_part3-7-320.jpg)
![Dictionaries
• Hash tables, "associative arrays"
• d = {"duck": "eend", "water": "water"}
• Lookup:
• d["duck"] -> "eend"
• d["back"] # raises KeyError exception
• Delete, insert, overwrite:
• del d["water"] # {"duck": "eend", "back": "rug"}
• d["back"] = "rug" # {"duck": "eend", "back":
"rug"}
• d["duck"] = "duik" # {"duck": "duik", "back":
"rug"}](https://image.slidesharecdn.com/2015bioinformaticsbiopythonpart3-151116134050-lva1-app6892/85/2015-bioinformatics-bio_python_part3-8-320.jpg)
![Regex.py
text = 'abbaaabbbbaaaaa'
pattern = 'ab'
for match in re.finditer(pattern, text):
s = match.start()
e = match.end()
print ('Found "%s" at %d:%d' % (text[s:e], s, e))
m = re.search("^([A-Z]) ",line)
if m:
from_letter = m.groups()[0]](https://image.slidesharecdn.com/2015bioinformaticsbiopythonpart3-151116134050-lva1-app6892/85/2015-bioinformatics-bio_python_part3-9-320.jpg)












![Calculation of Propensities
Pr[i|b-sheet]/Pr[i], Pr[i|-helix]/Pr[i], Pr[i|other]/Pr[i]
determine the probability that amino acid i is in
each structure, normalized by the background
probability that i occurs at all.
Example.
let's say that there are 20,000 amino acids in the database, of
which 2000 are serine, and there are 5000 amino acids in
helical conformation, of which 500 are serine. Then the
helical propensity for serine is: (500/5000) / (2000/20000) =
1.0](https://image.slidesharecdn.com/2015bioinformaticsbiopythonpart3-151116134050-lva1-app6892/85/2015-bioinformatics-bio_python_part3-23-320.jpg)









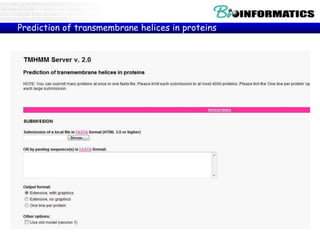



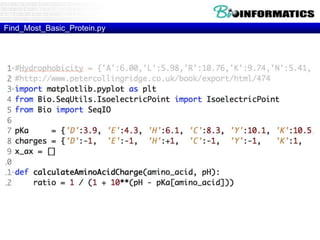
The document discusses various bioinformatics tools and concepts. It introduces GitHub as a code hosting platform and describes control structures, lists, and dictionaries in Python. It also covers topics like regular expressions, Biopython, parsing sequences from online databases, secondary structure prediction using Chou-Fasman algorithm, and transmembrane region prediction using Kyte-Doolittle hydropathy plots.


























































![Bio ontologies and semantic technologies[2]](https://cdn.slidesharecdn.com/ss_thumbnails/bioontologiesandsemantictechnologies2-180509123734-thumbnail.jpg?width=640&height=640&fit=bounds)




























