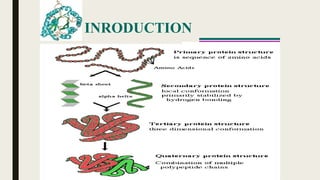
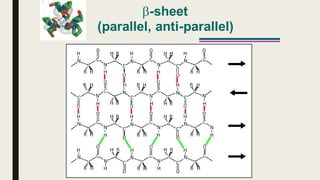
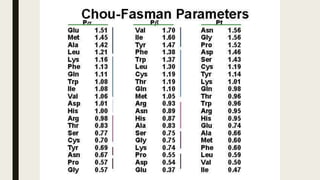
This document contains a presentation submitted by 5 students to their lecturer at Daffodil International University. It summarizes the Chou-Fasman method for secondary structure prediction of proteins. The method uses a statistical approach to assign probabilities for alpha helix, beta sheet, and coil conformations based on amino acid preferences. It then applies rules to scan the protein sequence and identify regions that meet the criteria for each secondary structure type. The accuracy of secondary structure prediction using this method is around 50-60%.