Downloaded 18 times


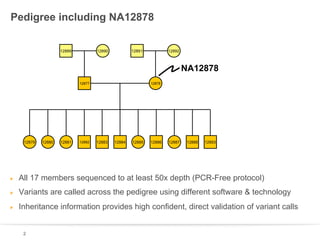
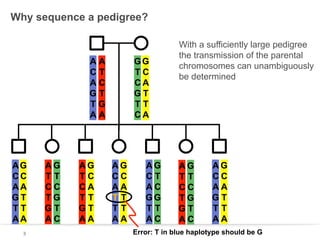
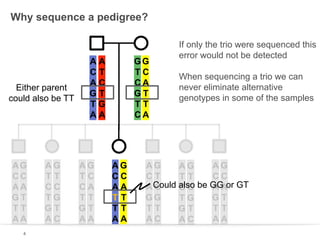
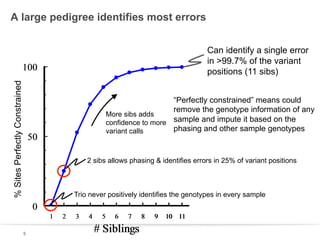
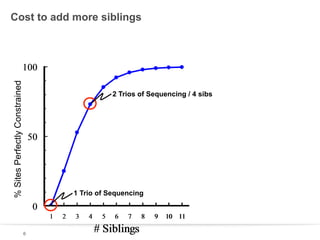


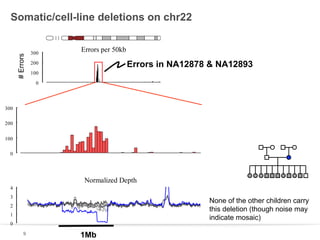
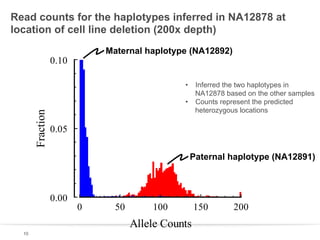
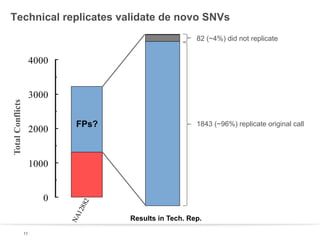


This document discusses sequencing a 17-member pedigree including individual NA12878 to 50x depth using different variant calling software and technologies. Sequencing a large pedigree allows unambiguous determination of parental chromosome transmission and identifies errors in over 99.7% of variant positions with 11 siblings. While adding more siblings increases confidence in variant calls and phasing, it also increases costs. Somatic deletions were found in NA12878 and another individual, but not carried by other children. Technical replicates validated over 96% of de novo SNV calls in NA12878. Moving forward, selecting pedigrees with multiple siblings and one high quality family over lower quality trios would provide the best reference, while long reads on one or two samples could
































![El asesinato del profesor de matemáticas [2] luis y carla. LEON CANO](https://cdn.slidesharecdn.com/ss_thumbnails/elasesinatodelprofesordematemticas2luisycarla-130321184124-phpapp01-thumbnail.jpg?width=640&height=640&fit=bounds)

































































