Download to read offline




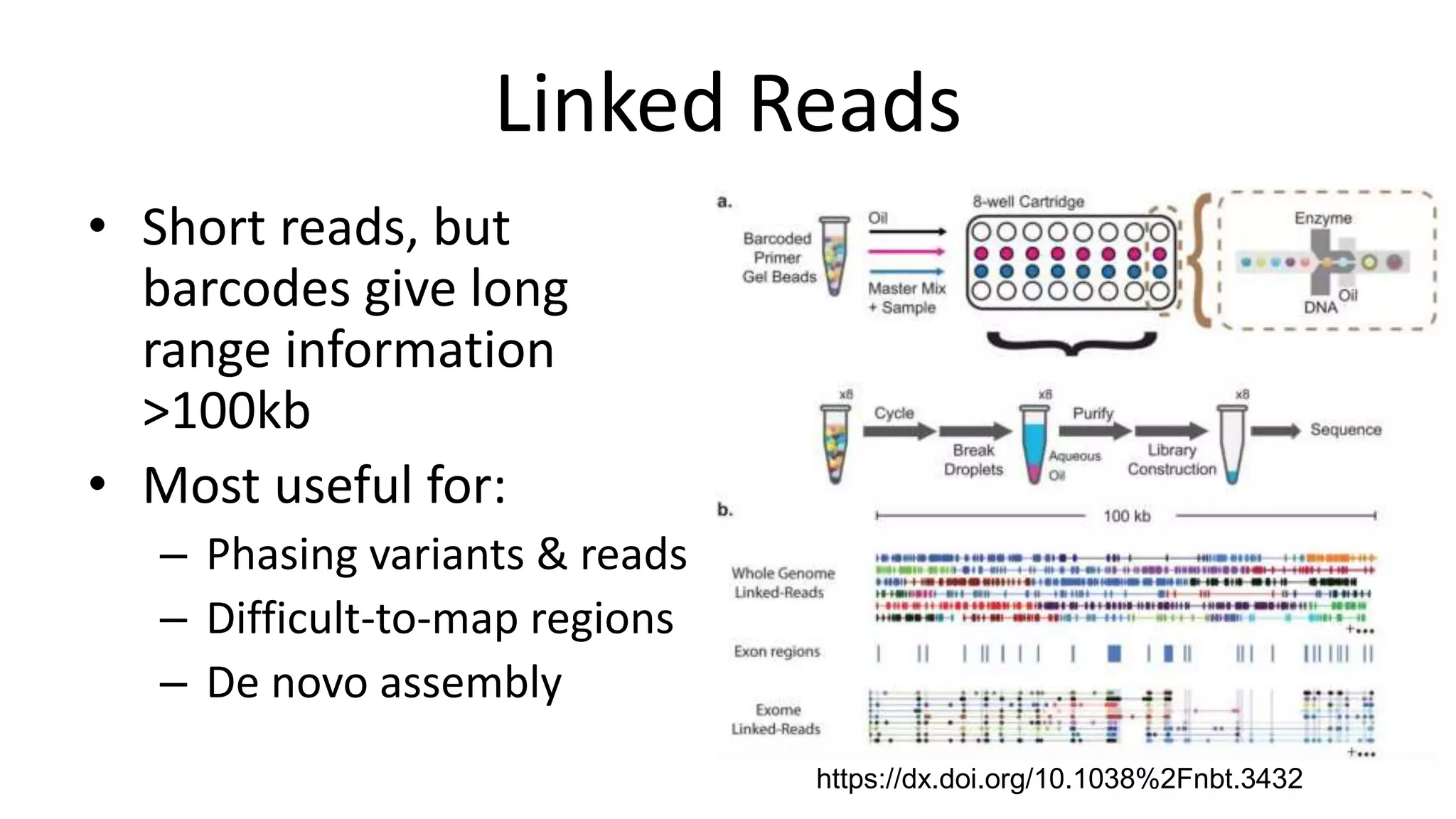
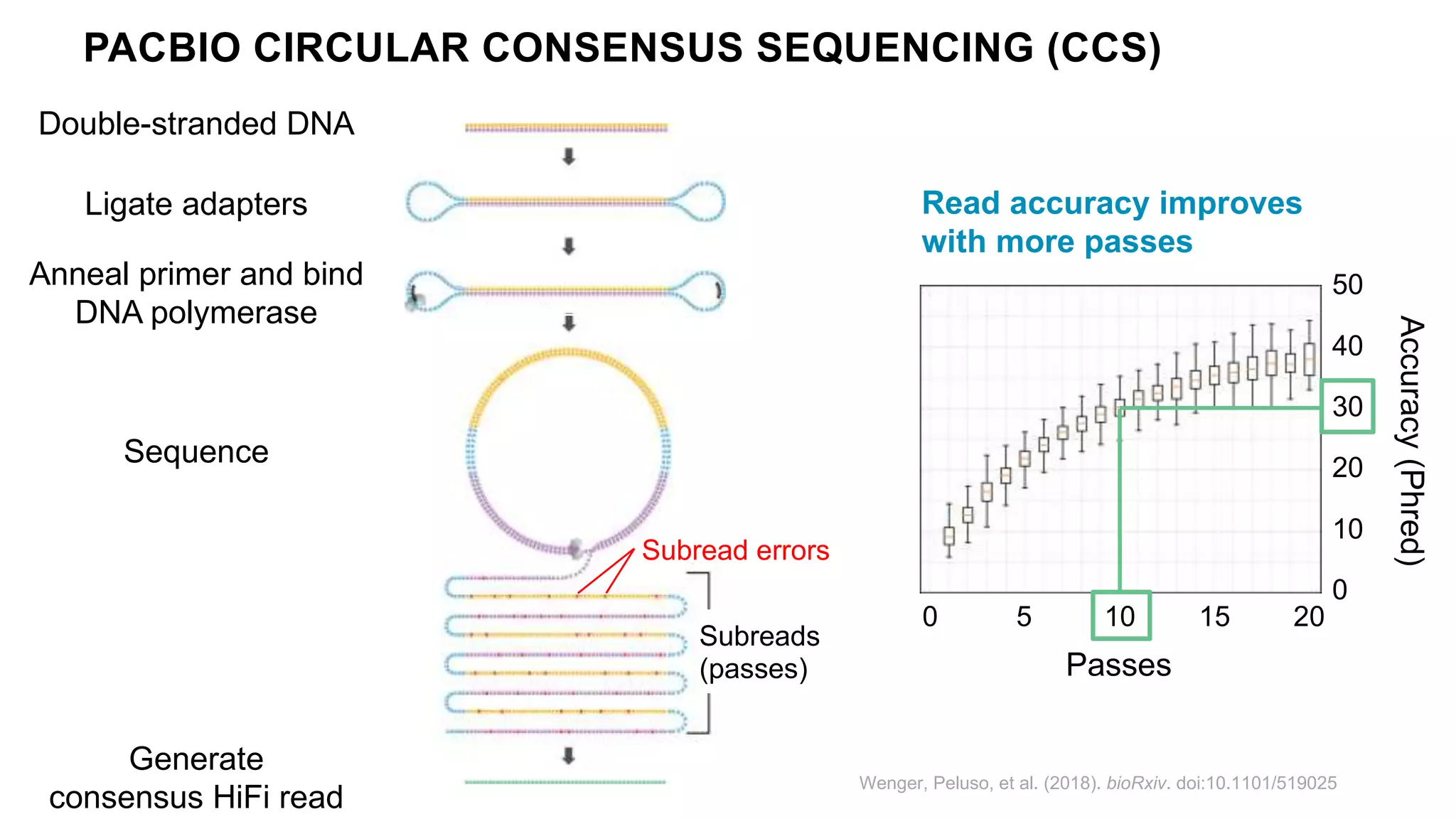
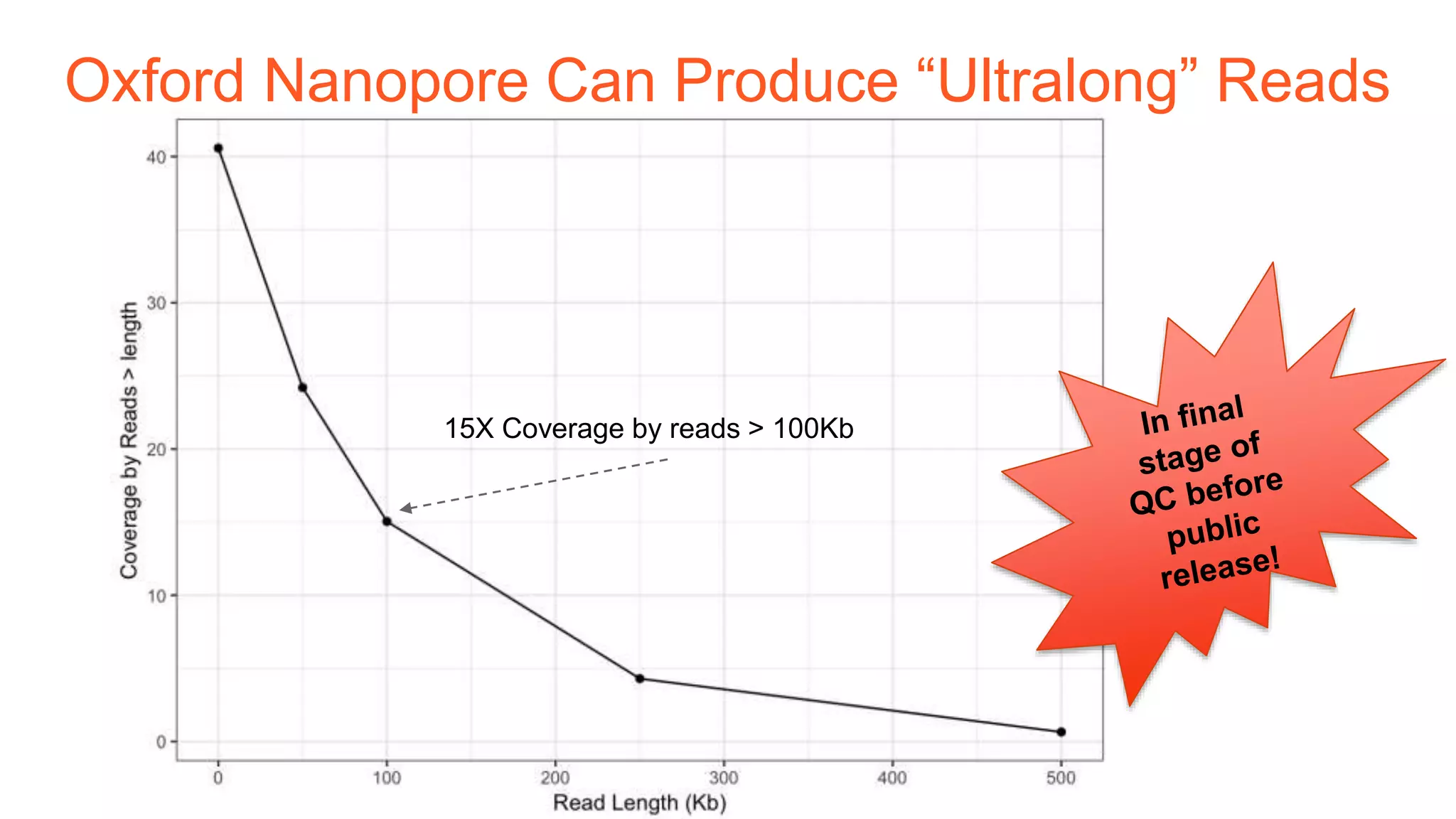

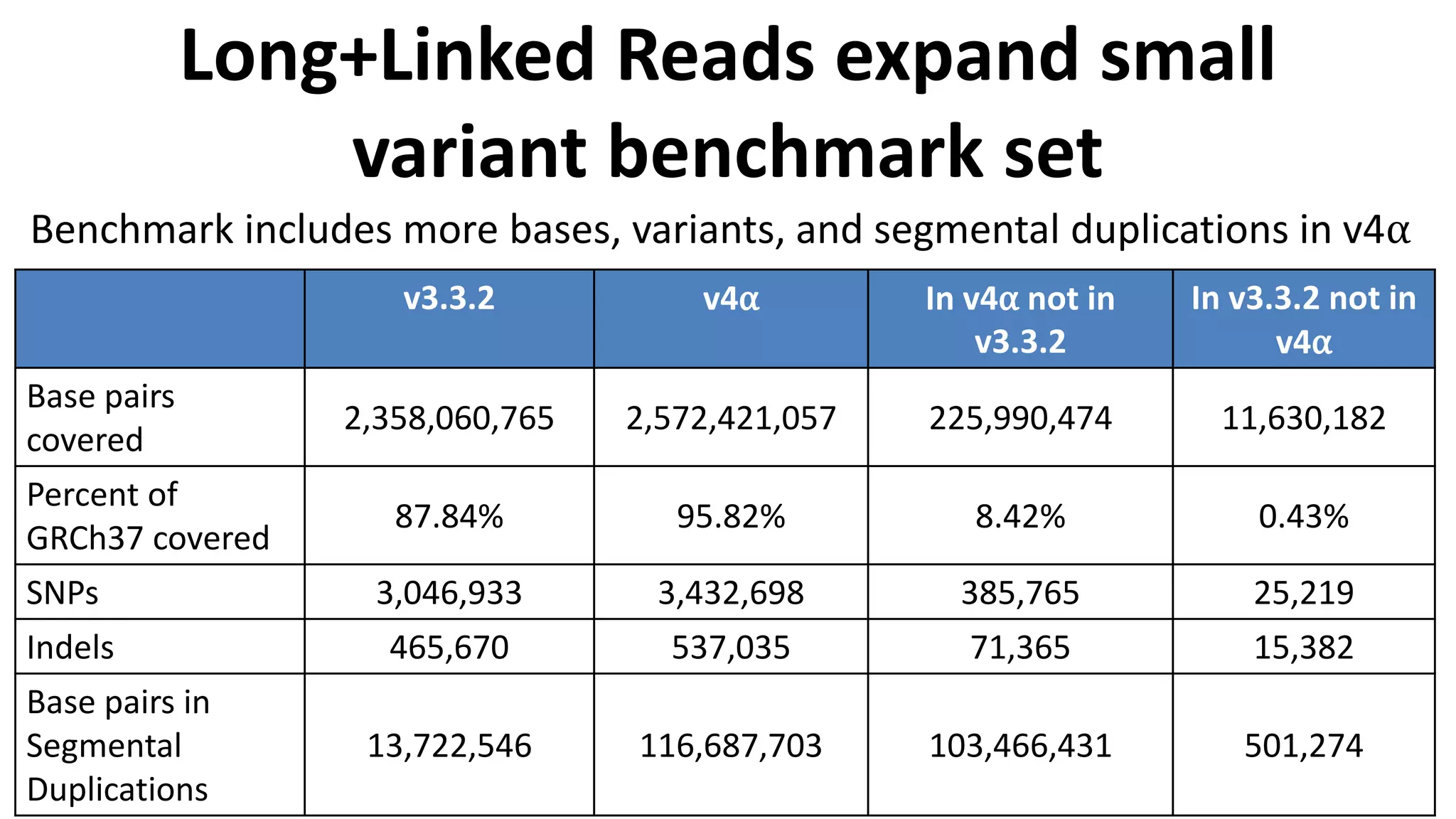
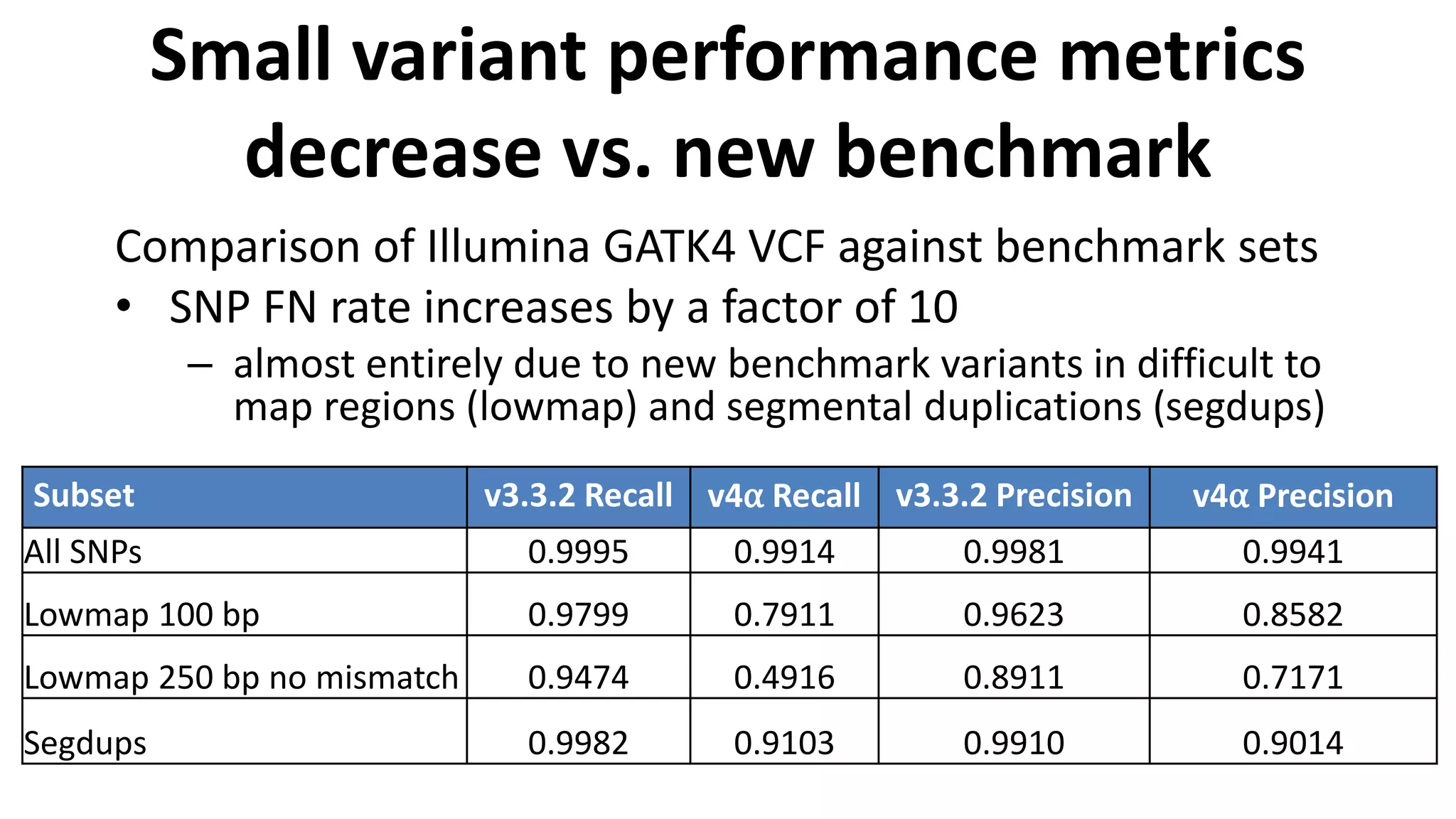
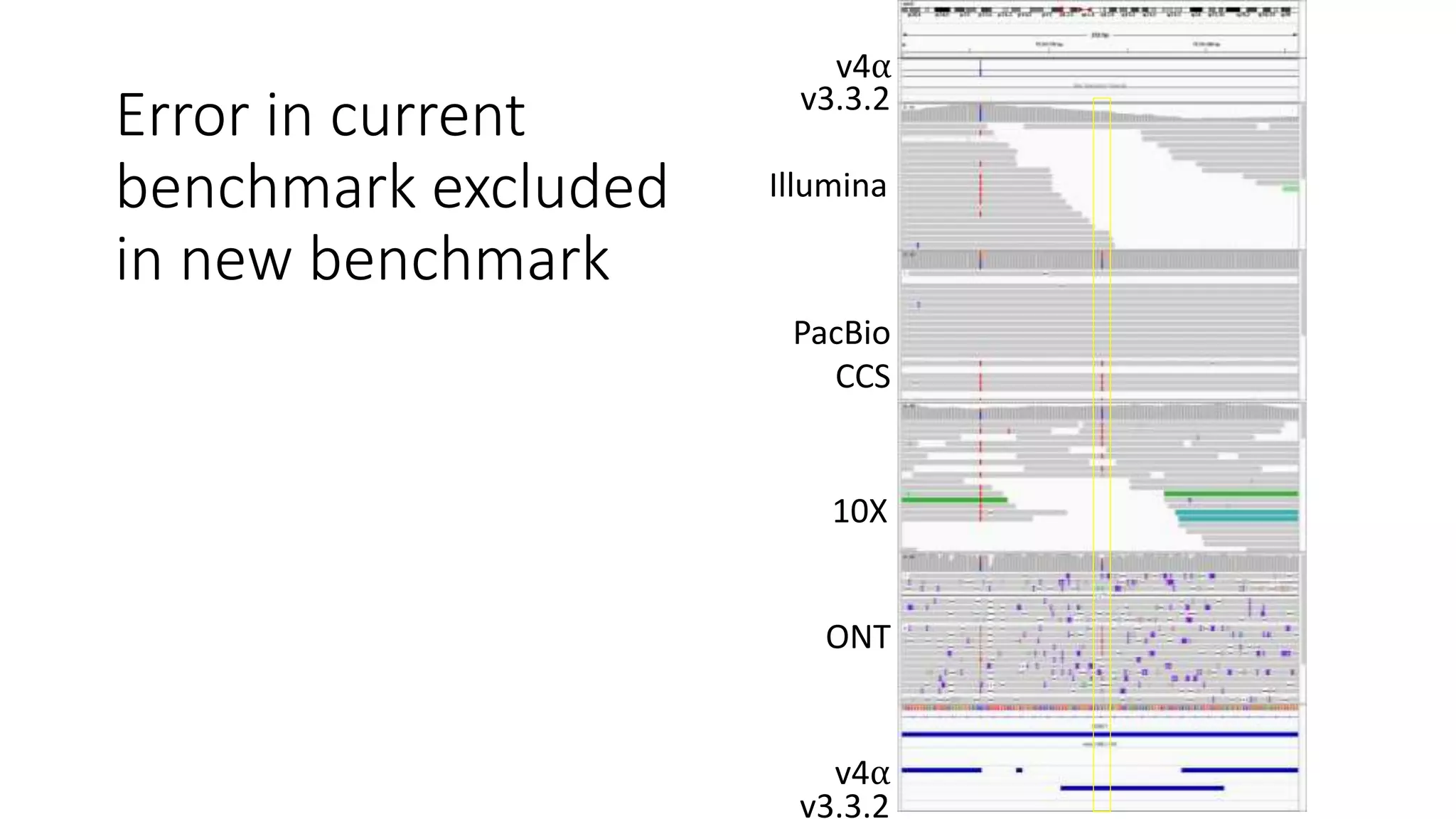



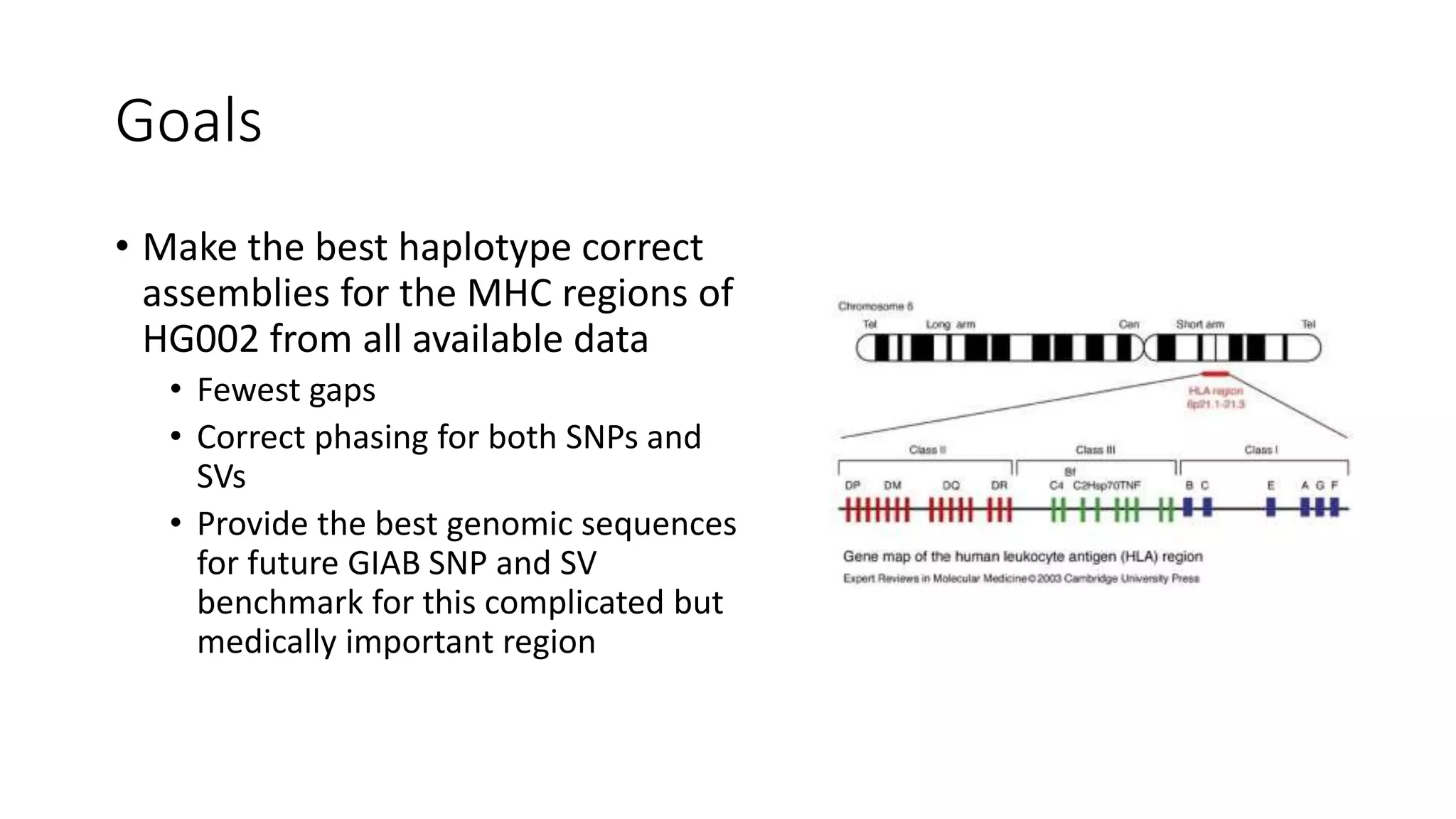
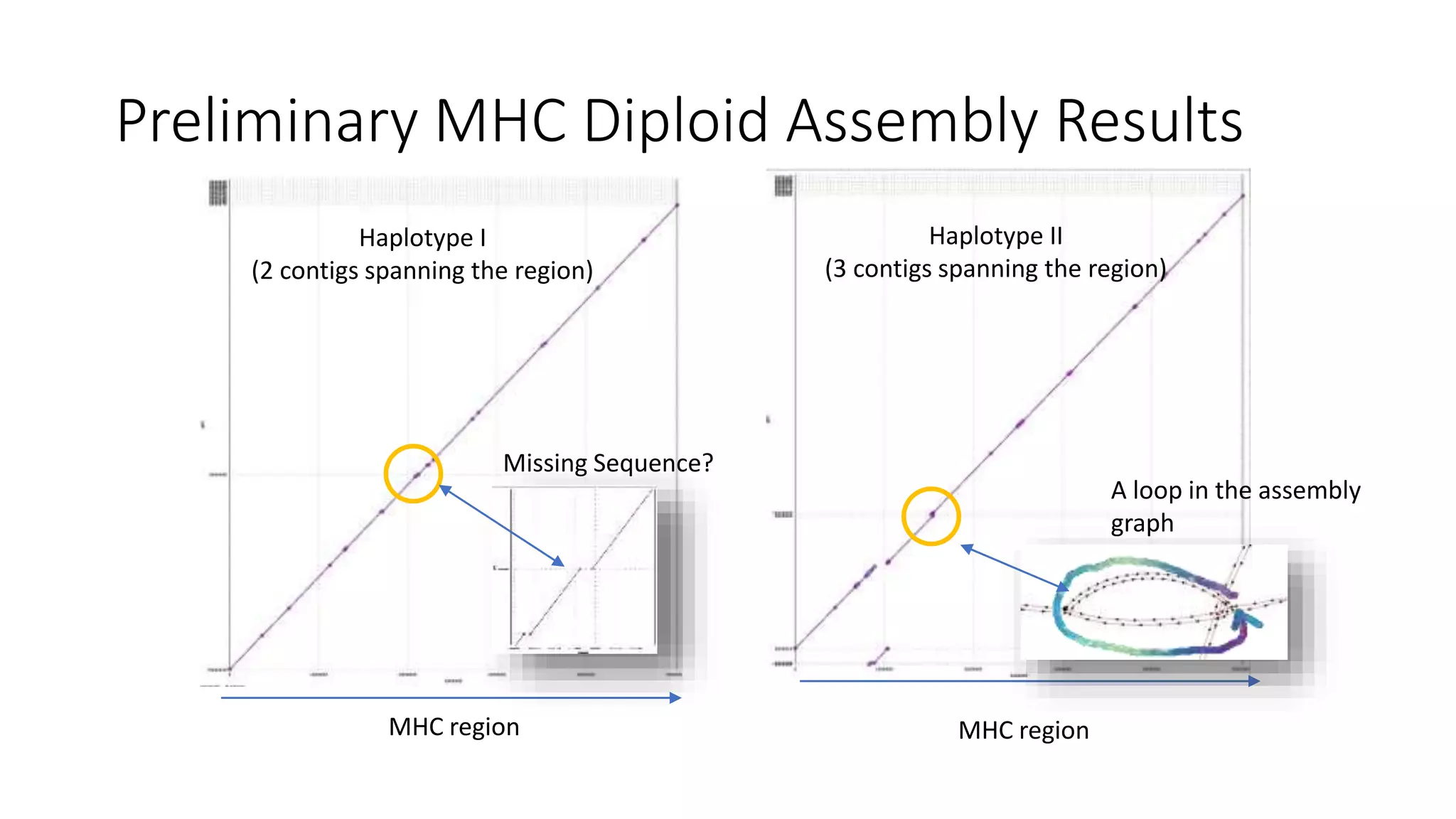
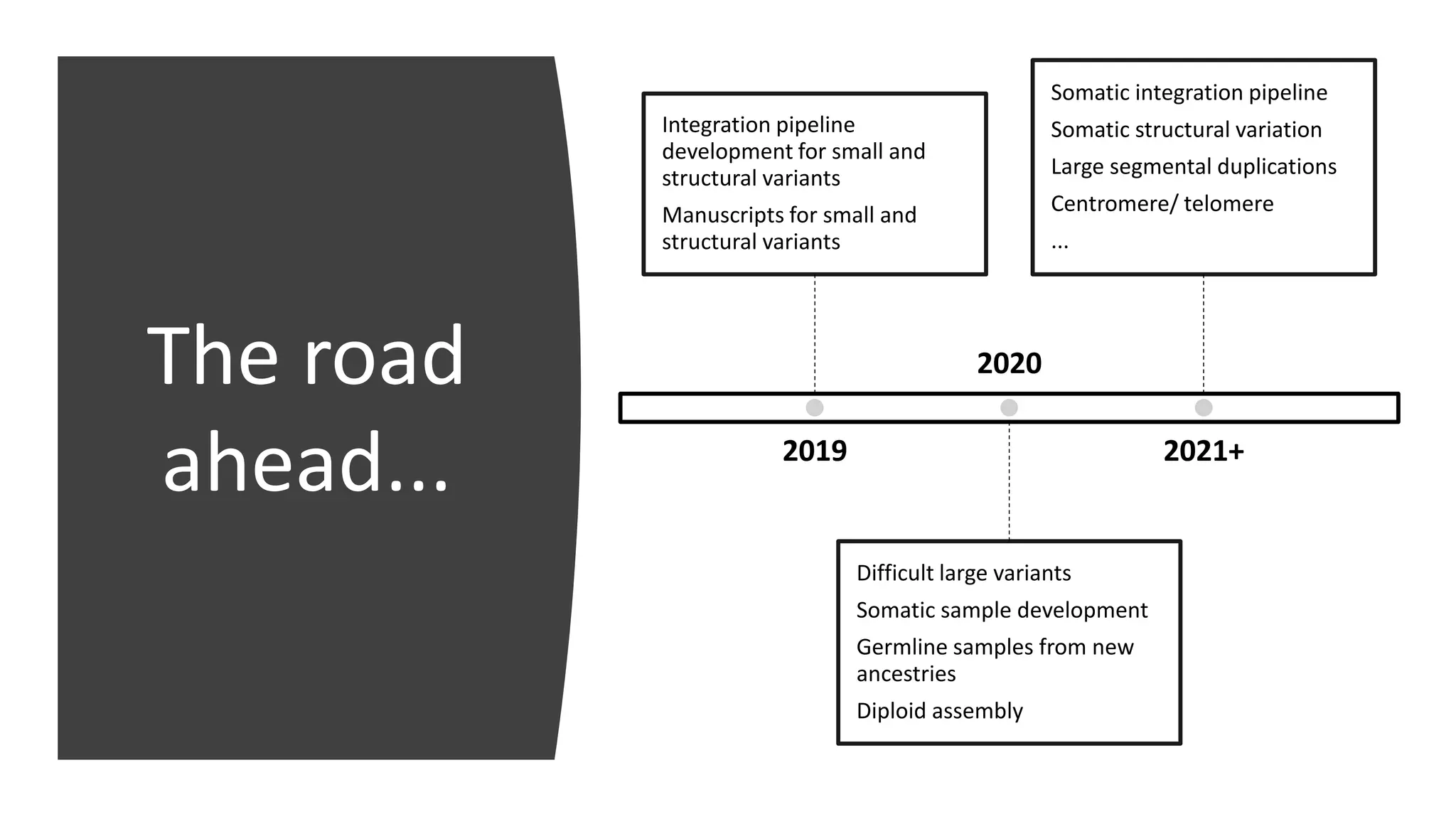



The Genome in a Bottle Consortium provides reference samples and genomic data to enable the development and benchmarking of genome analysis tools, and they have recently published new resources from linked and long read sequencing including 10x Genomics, PacBio, and Oxford Nanopore data, which have expanded their small variant benchmark set and enabled the development of a structural variant benchmark.











































































