Downloaded 201 times



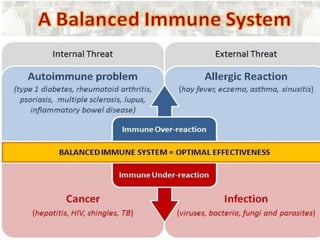
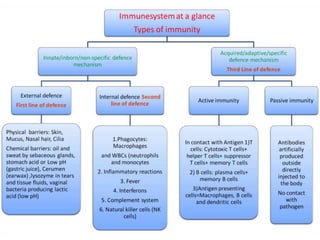
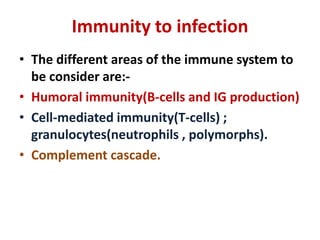




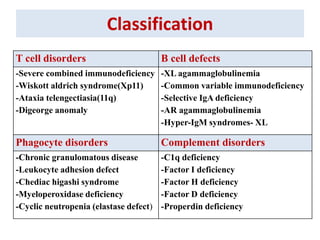
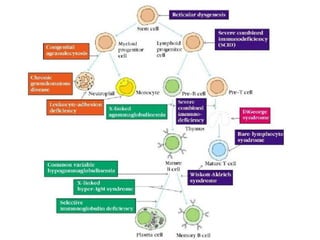
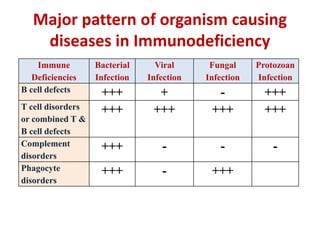




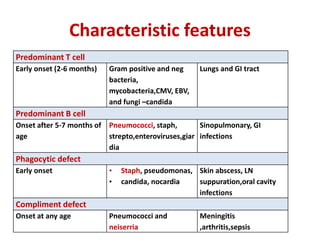


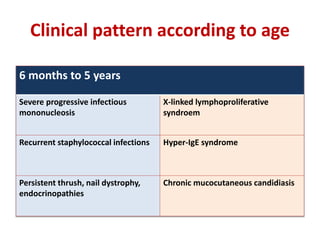



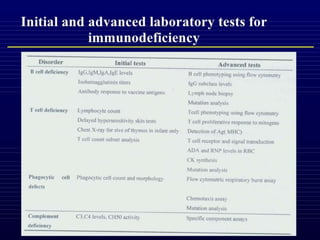



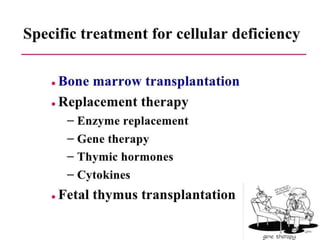





This document outlines immunology and immunity to infection. It discusses the immune system's role in fighting infection, including humoral immunity from B cells and cellular immunity from T cells. It also addresses intracellular and extracellular pathogens. The document then focuses on primary and secondary immunodeficiency, describing the types of primary immunodeficiencies including B cell, T cell, phagocytic, and complement defects. It provides guidance on clinical evaluation and diagnostic approach for patients with suspected immunodeficiency.











































































































