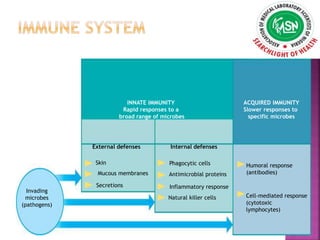
The document provides an overview of immunodeficiency disorders, categorizing them into primary and secondary types with details on symptoms, causes, and genetic factors. It discusses specific types of immunodeficiencies such as B-cell, T-cell, phagocytic, and complement deficiencies, as well as associated complications. Diagnosis and treatment options, including immunoglobulin therapy and transplants, are highlighted, emphasizing the importance of early intervention for improving patient outcomes.







![HYPOGAMMAGLOBULINEMIA
Characterized by low or deficient levels of any of
the immunoglobulins (immunoglobulin A [IgA],
IgE, IgG and IgG subclasses, IgM),
An abnormal response of immunoglobulins to
vaccinations
Ear and pulmonary infections are common;
gastrointestinal problems such as diarrhea,
malabsorption, and symptoms of irritable bowel
syndrome also occur in children who have
common variable immunodeficiency.
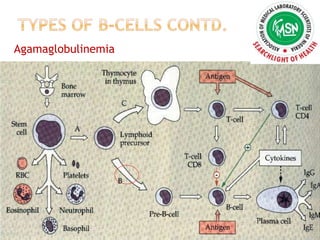
AGAMMAGLOBULINEMIA
Accounts for 13% of antibody disorders, with X
chromosome–linked Bruton tyrosine kinase
defect accounting for 84% of
agammaglobulinemias.
Complete absence of B cells in children’s
peripheral blood and in their umbilical cord blood.
Absence of tonsils or lymph nodes and Decrease
immunoglobulin subtypes.
(Gathmann et al., 2009)](https://image.slidesharecdn.com/jamesebi-170329204041/85/IMMUNODEFICIENCY-IN-HAEMATOLOGY-10-320.jpg)







































































![CASE_PRESENTATION_ON_subdural_hematoma(SDH)[1 FINAL PPT]-1.pptx](https://cdn.slidesharecdn.com/ss_thumbnails/casepresentationonsubduralhematomasdh1finalppt-1-260129172522-d405d375-thumbnail.jpg?width=640&height=640&fit=bounds)







