Downloaded 13 times




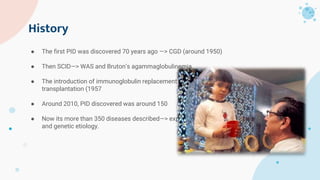

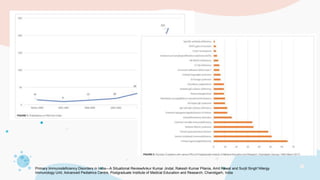







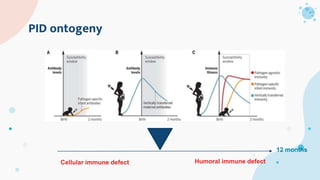



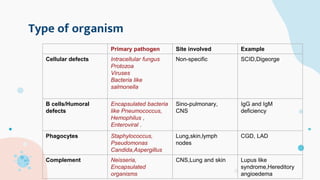

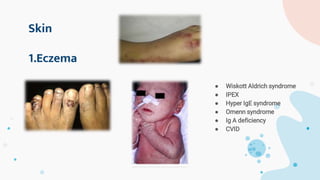
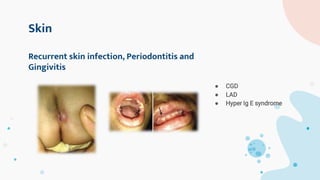
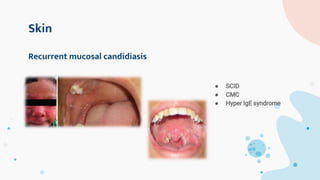
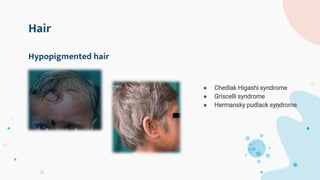
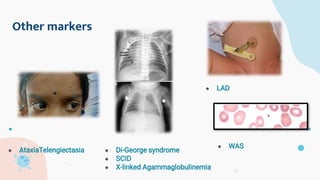




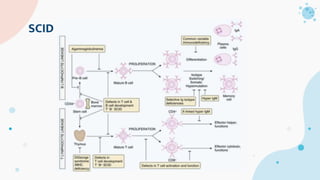
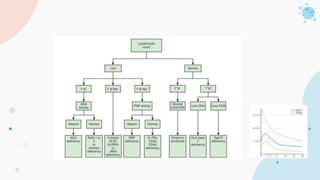


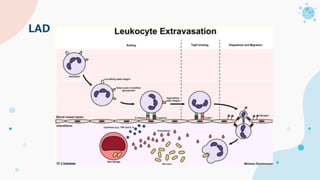

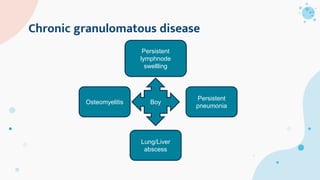


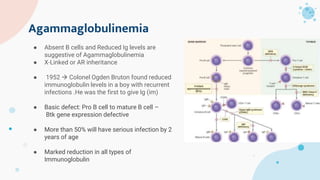
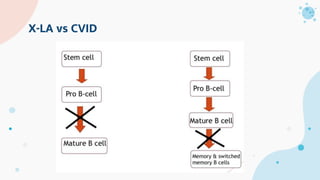






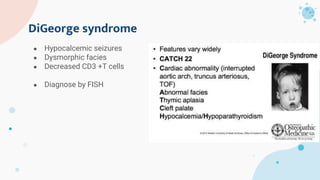

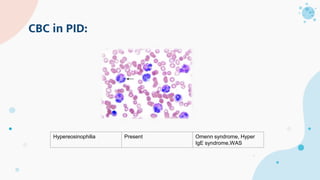

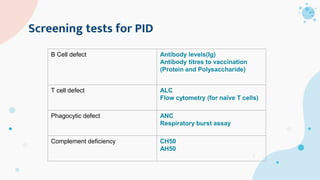
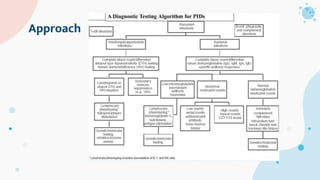


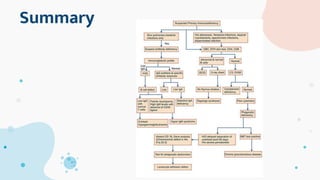


The document provides background information on primary immunodeficiency diseases (PIDs) and discusses their approach and management. It notes that over 350 PIDs have been identified so far. PIDs are classified based on the component of the immune system involved, such as combined antibody and T-cell deficiencies. Clinical markers for specific PIDs include recurrent infections, eczema, hair abnormalities, and organ involvement. Case scenarios demonstrate examples of SCID, LAD, CGD, agammaglobulinemia, and SCN. Screening tests and CBC findings are discussed to rule out certain PIDs. Management involves hematopoietic stem cell transplantation or IVIG replacement depending on the disorder.



















































































