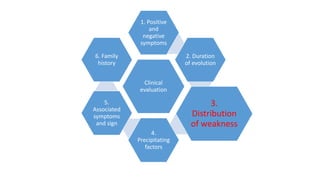
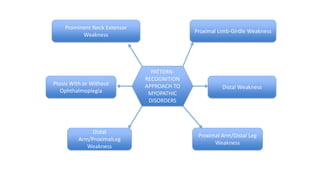
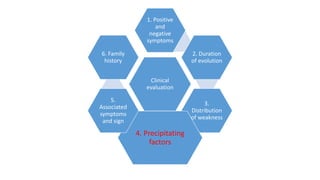
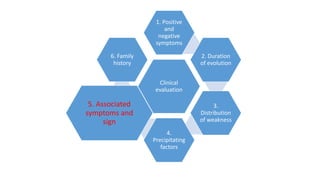
The document provides guidance on evaluating patients presenting with suspected muscle disease. It outlines the key goals of determining the site of lesion, cause, and available treatments. Symptoms like weakness, fatigue, myalgia and their patterns are discussed to help determine underlying conditions. The temporal evolution, including acute vs chronic onset and progression, helps differentiate between genetic, inflammatory and metabolic myopathies. Considering symptom precipitants and distributions can provide clues to diagnose specific myopathies based on pattern recognition of proximal, distal or other muscle group involvement.























![Myopathies Presenting in Adulthood
• Muscular dystrophies
– Limb-girdle
– Facioscapulohumeral
– Becker
– Emery-Dreifuss
• Inflammatory myopathies
– Polymyositis
– Dermatomyositis
– Inclusion body myositis
– Viral [HIV]
• Metabolic myopathies
– Acid maltase deficiency
– Lipid storage diseases
– Debrancher deficiency
– Phosphorylase b kinase deficiency
– Mitochondrial myopathies
• Endocrine myopathies
– Thyroid
– Parathyroid
– Adrenal
– Pituitary disorders
• Toxic myopathies
– Alcohol
– Corticosteroids
– Local injections of narcotics
– Colchicine
– Chloroquine
– Statins
• Myotonic dystrophy
• Distal myopathies
• Nemaline myopathy
• Centronuclear myopathy](https://image.slidesharecdn.com/otyhbzexqqk4ci06iqgh-signature-14d46799605c8398ccf53718c60b47b524e48d3b0496686c2da422d12af102e3-poli-180511192403/85/Approach-to-myopathy-26-320.jpg)