Downloaded 184 times

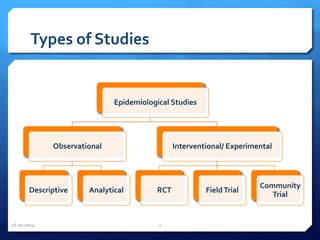

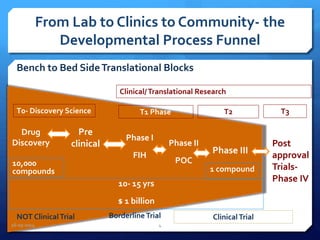



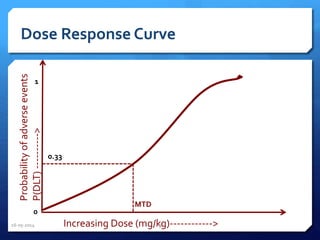












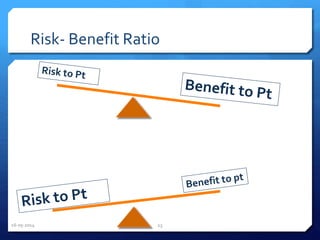

















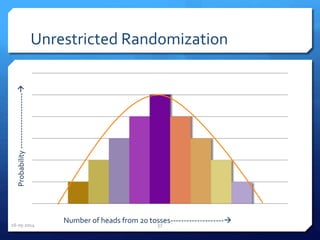
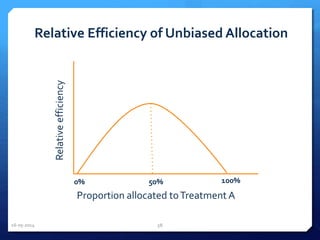





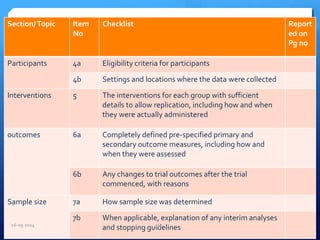
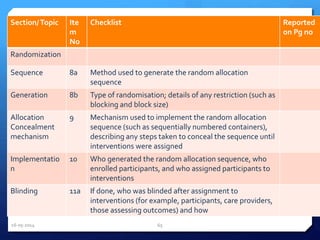



The document outlines the process and importance of clinical trials, detailing various types of studies, phases of trials, and ethical considerations involved in conducting research. It emphasizes the necessity of clinical trials registration for transparency and accountability, highlights the role of ethical guidelines, and discusses the methodology for designing and analyzing trials. Additionally, it provides a comprehensive overview of important components like informed consent, risk-benefit analysis, and statistical methods used in clinical research.

































































































