Hikaye
L.H., 14yaşında kadın hasta, mens görememe ve
meme gelişimi olmaması gibi sekonder seks
karakterlerinin gelişmemesi nedeniyle endokrinoloji
bölümüne başvuruyor. SGA doğmuş, iyi bir sağlığı
varmış ve zekası normalmiş. Fizik muayenede boyu
kısa, Tanner 1 seksüel gelişim, tahta göğüs ve meme
başı ayrık tespit ediliyor. Bunun üstüne doktoru FSH,
GH, kemik yaşı ve kromozom analizi istiyor. Sonuçta
normal GH, yüksek FSH ve anormal karyotip (45,X)
çıkıyor. Hastaya Turner Sendromu teşhisi konuluyor ve
lineer uzamayı sağlamak için GH başlanıyor. Bundan 1
yıl sonra, sekonder seksüel karakterlerin gelişmesi için
östrojen ve progesteron başlanıyor.
3.
Tanım
Turner Sendromu,kadınlarda, ikinci X
kromozomunun tamamen veya kısmen
eksikliğinden kaynaklanan (Female
Monosomy X), prenatal dönemde bulgu
vermeye başlayan, kısa boy, ovarian
disgenesis ve seksüel immatürite ile
prezente olan bir seks kromozomu
sendromudur.
4.
Etyoloji ve İnsidans
1/2000-5000 (canlı doğum)
45,X %50
45,X/46,XX %20
46,X,i(Xq) %15
46,X,r(X) %5
45,X,del(Xp) %5
Diğer %5
5.
Gametlerden birindenseks kromozomu
iletiminde bozukluk: 45,X karyotipin %70-
80’i spermden kaynaklı X kromozomu
eksikliği
Seks kromozomlarından birinin zigot veya
erken embriyo zamanında kaybı: 45,X
mosaizm’i
Patogenez
Oosit gelişimiiçin bir tane X kromozomu
yeterli olurken, oosit süregelimi ve
devamlılığı iki tane X kromozomu
gerektirmektedir.
İkinci X kromozomu eksikliğinde;
oosit dejenerasyonu ovarian atrofi
streak fibrous gonad (yüksek FSH)
8.
Patogenez
Kistik higroma,lenfödem, tahta göğüs,
kardiak, renal ve işitsel anomaliler, tam
olarak tanımlanmamakla birlikte,
tamamen inaktivasyona gitmeyen X gen
bölgeleriyle ilgili oldukları öne
sürülmektedir.
9.
Fenotip ve DoğalÖykü
Tüm gebeliklerin %1-2’si 45,X karyotiple
sonuçlanır, bunların %1’inden azı canlı
doğumla sonuçlanır.
Düşükler ve zamanlamalarına
bakıldığında ikinci seks kromozomu
intrauterin hayatta kalmada özellikle ikinci
trimester’in başında etkili ve gerekli
olmaktadır.
10.
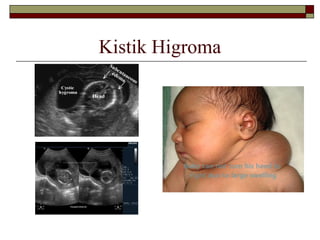
Fenotip ve DoğalÖykü
Prenatal bulgular;
-Lenfatik drenaj sistemin matürasyonunda
bozukluk: Lenfödem
-Fetal USG’de kistik higroma, hidrops fetalis
11.
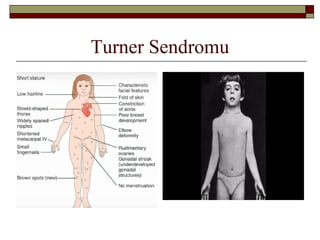
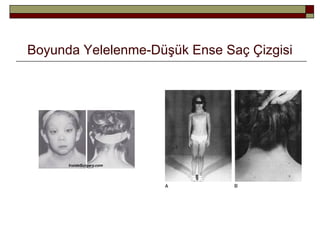
Fenotip ve DoğalÖykü
Doğumdaki bulgular;
-Boyunda yelelenme (webbed neck)
-El ve ayak sırtında ödem
-Renal anomaliler
-Kardiyovasküler anomaliler
Fenotip ve DoğalÖykü
Çocukluk ve adölesan bulguları;
-Zeka normal (yapısal anomaliler !)
-Sensörinöral işitme kaybı
-Uzaysal algı, ince motor uygulamalarında
zayıflık
-Aortik kök dilatasyonu ve disseksiyonu
-Kubitus valgus
14.
-Ovaryan disgenezis;
-Streak fibrousgonadlar
-%10-20 spontan pubertal gelişme (meme
gelişimi, pubik kıllanma)
-%2-5 spontan menstrüasyon
-%100’e yakın infertilite
15.
Ayrıca;
-Tiroidit (hipotiroidizm)
-İnflamatuarbarsak hastalığı
-Östrojen defekti;
-Osteoporoz ve osteoporotik kırıklar
-Atheroskleroz
-İskemik kalp hastalığı
-Stroke
-Diabetes mellitus (Tip 1-2)
16.
Kromozom Yapısı veKlinik
Özellikler
i(Xq) vakaları klasik 45,X vakalarına
benzer,
del(Xp)’de kısa boy ve konjenital
malformasyonlar
del(Xq)’da sıklıkla sadece gonadal
disfonksiyon görülür.
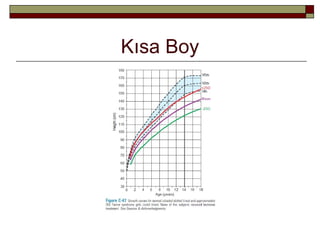
Yönetim
5 persentilinaltı genellikle kemik yaşı
15’e ulaşana kadar GH ile tedavi
edilmelidir.
-Beklenti; final boyda 10cm’lik artıştır ancak
östrojen tedavisi GH tedavisinin etkisini
azaltmakta ve bu artış beklenenden daha az
olmaktadır.
25.
Yönetim
14-15 yaşlarındasekonder seks
karakterlerinin gelişimi ve osteoporoz
başta olmak üzere östrojen defektinden
kaynaklanan komplikasyonları önlemek
için östrojen tedavisi başlanmalı.
Östrojen tedavisinin 2. yılında
menstrüasyonu indüklemek için tedaviye
progesteron eklenmeli, ancak..
26.
Yönetim
Östrojen veprogesteron tedavisinin
tromboz indükleyici etkileri Turner
Sendrom’u olan hastalarda; normal
populasyonda hormon tedavisi alanlara
göre daha yüksek riskli prezente
olabilmektedir.
27.
Yönetim
Aortik kökdilatasyonu ve valvar kalp
hastalıkları için seri ekokardiyografi takibi,
Konjenital anomaliler için renal USG,
Diabet tespiti için glukoz tolerans testi.
28.
Yönetim
Yeterli kardivaskülerve renal fonksiyonu
olan hastalar in vitro fertilizasyon veya
ovum donation’la gebe kalabilirler,
ancak..
-Gebelikle birlikte yüksek oranda artmış
aort disseksiyonu ve rüptürü riski ile karşı
karşıya kalırlar.