Downloaded 55 times




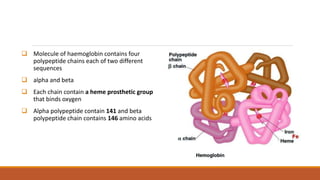
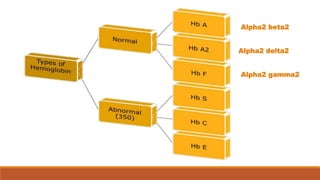
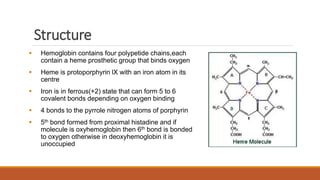
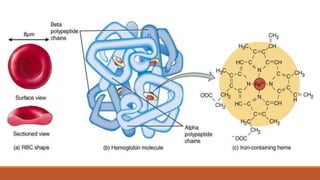
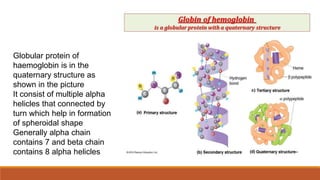



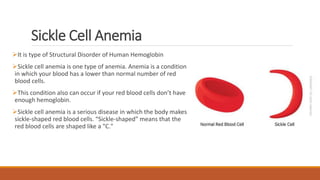
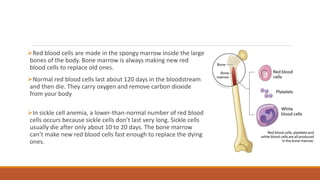

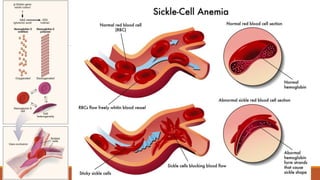

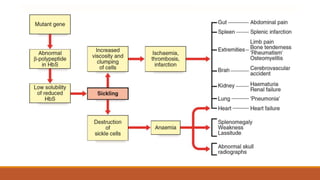
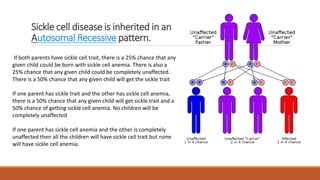
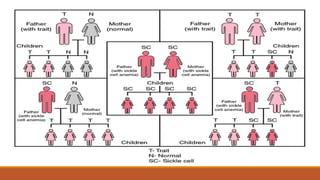




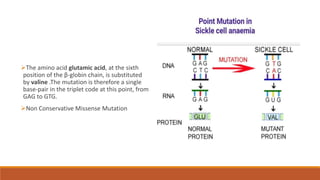

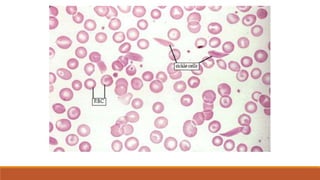
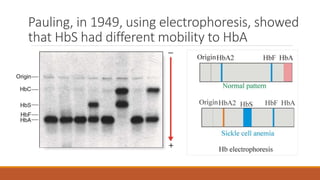





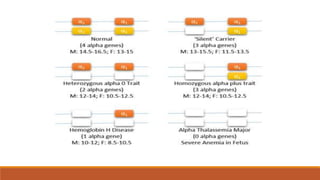

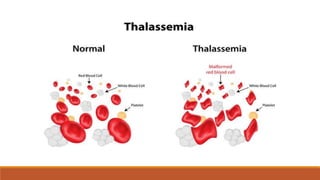

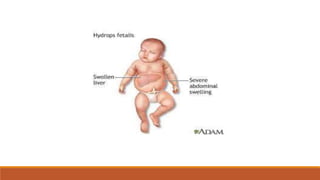







Hemoglobinopathies are disorders of hemoglobin structure or synthesis. The document discusses hemoglobin structure and function, as well as two main types of hemoglobinopathies: sickle cell anemia and thalassemia. Sickle cell anemia is a structural disorder caused by a mutation resulting in abnormal hemoglobin S, which causes red blood cells to sickle and break down early. Thalassemia is a disorder of hemoglobin synthesis, where there is reduced or absent alpha or beta globin chain production leading to anemia. Common symptoms, inheritance patterns, and treatments are described for both conditions.



































































