3
04/03/2025 05:02 PM
TheHistory of Thalassemia
• Thalassemia, or Mediterranean anaemia, was first described in 1925 by a Detroit
physician who studied Italian children with severe anaemia.
• It was until 1946, that the cause of thalassemia was found to be an abnormal
haemoglobin structure.
• It was derived from the Greek word thalassa (θάλασσα), "sea“ and New Latin -
emia (from the Greek compound stem -aimia (-αιμία), from haima (α μα), "
ἷ blood").
• It was coined because the condition called "Mediterranean anaemia" was first
described in people of Mediterranean ethnicities.
• "Mediterranean anaemia" was renamed thalassemia major once the genetics
were better understood.
4.
4
04/03/2025 05:02 PM
Whatis a Thalassemia?
• Thalassemia results from a reduced rate of synthesis of normal α globin chains
‐
or β globin chains (the α and β thalassemias).
‐ ‐ ‐
• Thalassemia has an autosomal recessive pattern of inheritance.
• Autosomal means that the gene in question is located on one of the numbered,
or non-sex chromosomes.
• Recessive disorder is where two copies of the mutation are needed to cause the
disease.
• Dominant means that a single copy of the disease-associated mutation is
enough to cause the disease.
5.
5
04/03/2025 05:02 PM
Whatis a Thalassemia?
• Autosomal recessive is one of several ways that a trait disorder or the
disease can be passed down through families and requires two copies of
the abnormal genes to be present in order for the disease or the trait to
develop.
• A genetic condition can occur when the child inherits one copy of a mutated
(changed) gene from each parent.
• The parents of a child with an autosomal recessive condition usually do not
have the condition.
6.
6
04/03/2025 05:02 PM
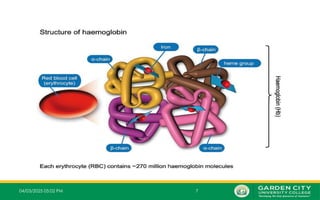
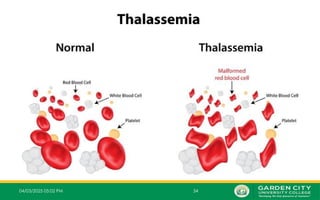
Whatis a Thalassemia?
• A thalassemia is a group of inherited blood disorders that can be passed from parents to their
children and affect the amount and type of Hb the body produces.
• People with thalassemia have one or more genetic mutations that they have inherited and that
result in a decreased production of normal Hb.
• One portion of Hb called heme is the molecule with iron at the center. Another portion is made of
up four protein chains called globin. Each of the four globin chains holds a heme group
containing one iron atom.
• Depending on their structure, the globin chains are designated as alpha, beta, gamma, or delta.
• Different types of Hb are classified according to the type of globin chains they contain.
• The type of globin chains present is important in Haemoglobin’s ability to transport oxygen
8
04/03/2025 05:02 PM
•The cause of thalassaemia is an inherited (genetic) change, involving the genes which tell
the body how to make an important component in the RBC called Hb.
• Hb is made out of different parts. The main parts are the alpha chains and beta chains
which are put together to make the Hb molecule.
• In thalassaemia, part of the Hb is reduced - usually either the alpha chains or the beta
chains. This means that some of the Hb may not work properly. As a result, there is not
enough normal Hb and the RBCs break down easily leading to anaemia with various
symptoms. As such, the body tries to make more RBCs and more Hb. So, the blood system
goes into overproduction mode which can cause more symptoms and complications.
• Depending on the type of thalassaemia, the amount of the abnormal Hb varies. It can be
most of the body’s Hb, or only a small proportion. This is mainly what determines how
severe is the thalassaemia.
• There are also other individual factors involved. So, two people with the same type of
thalassaemia may have a different severity of illness from the same condition.
9.
9
04/03/2025 05:02 PM
Severityof thalassaemia
• Each type of thalassaemia (alpha and beta) is then classified into more types,
according to how severe the condition is. This mainly depends on how many
thalassaemia genes are involved.
• The mild types are called thalassaemia trait (or thalassaemia minor).
• The more severe beta types are;
Beta thalassaemia major (Cooley’s anaemia)
Beta thalassaemia intermedia.
• The more severe alpha forms are;
Hb Barts (very severe)
Hb H disease (moderate).
• There are also some rarer types of thalassaemia such as delta beta thalassaemia, or
combinations of a beta-thalassaemia gene with another abnormal Hb gene such as Hb
E.
10.
10
04/03/2025 05:02 PM
ThalassemiaSyndromes
• These syndromes are usually a result of deletions of one or more genes, although
approximately 20% of the mutations described are non-deletional.
• Clinically the main syndromes are;
Transfusion dependent thalassemia (Thalassemia Major)
Non Transfusion dependent thalassemia (Thalassemia Intermedia) with a
‐
moderate degree of anaemia due to a variety of genetic defects
Thalassemia minor, usually due to a carrier state for α‐ or β‐thalassemia
11.
11
04/03/2025 05:02 PM
Classification
•The are two main types of thalassemia depending on which part of globin chain is
produced in reduced amounts
i. Alpha Thalassemia
ii. Beta Thalassemia
13
04/03/2025 05:02 PM
α‐ThalassemiaSyndromes
• In alpha thalassemia, the Hb does not produce enough alpha protein.
• To make alpha-globin protein chains we need four genes, two on each chromosome
16; that is two from each parent.
• Alpha thalassemia will result if one or more of these genes is missing.
• The clinical severity of thalassemia depends on how many genes are
o Faulty
o Deleted
o Mutated
o Inactive.
14.
14
04/03/2025 05:02 PM
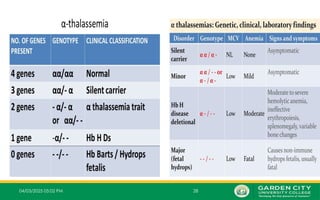
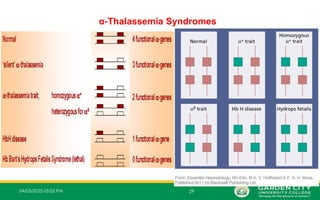
Typesof α‐Thalassemia Syndromes
• There are 4 different types of α-thalassemia syndromes;
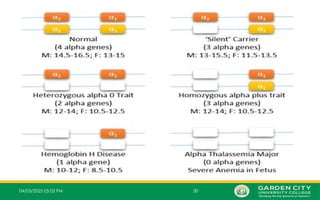
i. Alpha thalassemia silent carrier (1 mutated gene)
ii. Alpha thalassemia carrier (2 mutated genes)
iii. Alpha thalassemia Intermedia (3 mutated genes)
iv. Alpha thalassemia major (4 mutated genes)
15.
15
04/03/2025 05:02 PM
1.One Mutated Gene
– This type of thalassemia is characterized by inheritance of 3 normal α-
genes (-α/α α).
– These patients are referred to clinically as silent carrier of alpha
thalassemia.
– Other names for this condition are
Alpha thalassemia minima
Alpha thalassemia-2 trait
Heterozygosity for alpha (+) thalassemia minor.
16.
16
04/03/2025 05:02 PM
1.One Mutated Gene
– The affected individuals exhibit no abnormality clinically and may be
haematologically normal or have mild reductions.
– This condition generally causes no symptoms or signs of anaemia and will
not need treatment because the lack of alpha protein is so small that the Hb
functions normally.
– It is called "silent carrier" because it is difficult to identify its status by the
standard haematological studies. They are detected only by DNA Studies.
– A healthy person who has a child with symptoms of thalassemia is a carrier.
17.
17
04/03/2025 05:02 PM
2.Two Mutated Genes
– These are α+
thalassemia traits that are caused by loss of one or
‐
two globin genes on a single chromosome and are usually not
associated with anaemia.
– The patient has mild anaemia.
– It is known as alpha thalassemia carrier or traits.
– When parents are carriers, there is a one in 4 four, or 25 percent,
chance with each pregnancy to have a baby with alpha thalassemia
major.
18.
18
04/03/2025 05:02 PM
2.Two Mutated Genes
– Inheritance of 2 normal alpha genes due to;
• Heterozygosity for alpha (+) thalassemia (-α /- α) (one from each of the two
chromosomes) called a "trans deletion"
• Homozygosity for alpha (+) thalassemia (α α/--) (two on the same
chromosome) called a "cis deletion"
• All these will result in the development of alpha thalassemia carrier or alpha
thalassemia-1 trait.
19.
19
04/03/2025 05:02 PM
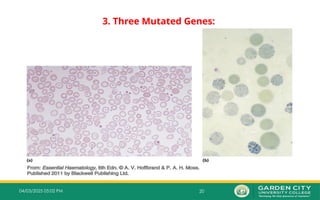
3.Three Mutated Genes:
– Inheritance of one normal alpha gene (-α/--) results in abundant formation of Hb H composed
of tetramers of excess beta chains.
– Hb H disease is usually with three genes absent or defective
– This condition is known as Hb H disease or Alpha Thalassemia Intermedia.
– The patient with Hb H disease is a type of chronic anaemia and they will need regular blood
transfusions throughout their life.
– Three α gene deletion leads to;
Moderately severe anaemia (Hb 7.0–11.0g/dL)
Microcytic hypochromic anaemia
Splenomegaly
– Hb H can be detected in red cells of these patients by;
Hb electrophoresis
Reticulocyte preparations using a supravital staining
21
04/03/2025 05:02 PM
3.Three Mutated Genes
– Infants born with alpha thalassemia intermedia appear
normal at birth but often develop anaemia and splenomegaly
by the end of their first year.
– Hepatomegaly is not a common finding but there may be
some association with mental retardation.
– The severe imbalance between the alpha chain production
(now powered by one gene, instead of four) and beta chain
production (which is normal) causes an accumulation of beta
chains inside the RBCs.
22.
22
04/03/2025 05:02 PM
3.Three Mutated Genes
– Normally, beta chains pair only with alpha chains.
– With three-gene deletion of alpha chains, beta chains will however, begin to
associate in groups of four, producing abnormal Hb called Hb H.
– Hb H therefore has two major problems;
First, the Hb H does not carry oxygen properly, making it functionally useless to the cell.
Second, the Hb H protein damages the membrane that surrounds the red cell, accelerating
cell destruction.
– The combination of the very low production of alpha chains and destruction
of red cells in Hb H disease produces a severe, life-threatening anaemia.
– Untreated, most patients die in childhood or early adolescence
23.
23
04/03/2025 05:02 PM
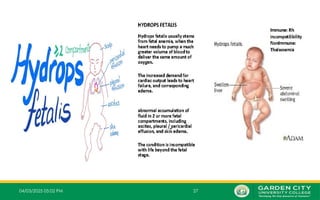
4.Four Mutated Genes
– This is alpha thalassemia major and is the most severe form of alpha
thalassemia, where all the four genes are absent or severely defective.
– It is known to cause Hb Bart’s hydrops fetalis, a serious condition in
which fluid accumulates in parts of the fetus’ body.
– The gamma chains produced during fetal life associate in groups of
four to form an abnormal Hb called Hb Bart.
– This is also called α0
thalassemia or alpha thalassemia major, where
the chromosome has no functional α genes.
24.
24
04/03/2025 05:02 PM
4.Four Mutated Genes
– Loss of all the four genes completely suppresses α chain synthesis and because
‐
the α chain is essential in fetal Hb as well as in adult Hb, this is incompatible with
‐
life and leads to death in utero (hydrops fetalis).
– It is also called Hb Bart’s hydrops fetalis because it was discovered at St
Bartholomew's Hospital in London, often abbreviated to Barts
– In this condition, there are no alpha genes in the individual's DNA, which causes
four gamma globins produced by the fetus to form abnormal Hb called Hb Bart.
– The symptoms of anaemia occur within the first trimester.
– Most individuals with this condition die before or shortly after birth.
25.
04/03/2025 05:02 PM25
4. Four mutated genes
– More than 20 different genetic
mutations that result in this
functional deletion of both pair of α-
globin genes have been identified.
– These individuals with this disorder
are unable to make any functional
Hb A, F, or A2.
• A fetus with four mutated genes
cannot produce normal Hb and is
unlikely to survive, even with
blood transfusions.
• Alpha thalassemia is common in
southern China, Southeast Asia,
India, the Middle East, and Africa.
26.
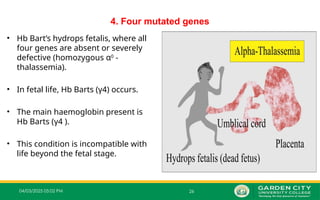
04/03/2025 05:02 PM26
4. Four mutated genes
• Hb Bart’s hydrops fetalis, where all
four genes are absent or severely
defective (homozygous α0
‐
thalassemia).
• In fetal life, Hb Barts (γ4) occurs.
• The main haemoglobin present is
Hb Barts (γ4 ).
• This condition is incompatible with
life beyond the fetal stage.
04/03/2025 05:02 PM31
Laboratory diagnosis α‐Thalassemia Syndromes
• In Hb H disease where there are
three α‐globin gene deletion
– (a) The blood film shows marked
hypochromic microcytic red cells
with target cells and poikilocytosis.
– (b) A supravital staining with
brilliant cresyl blue reveals multiple
fine deeply stained deposits (‘golf
ball’ cells) caused by precipitation
of aggregates of β‐globin chains.
• Hb H can also be detected as a
fast‐moving band on Hb
electrophoresis






































![THALASEMIA definition and pathophysiologyd].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/thalasemia-240515044127-8ce804c5-thumbnail.jpg?width=640&height=640&fit=bounds)












































