탈리도마이드 복용으로 인한대규모 약화사
고
부작용
보고
약물역학
연구
(환자군
연구)
사지결손증
: 팔, 다리가 비정상적
으로 짧은 기형
• 임신 첫 3-8주 복
용 시 예외없이 기
형아 출산
• 독일 약 2,500명,
세계적 10,000명
이상 발생 추정
약물 시판
중단
탈리도마이드
시판중지(1961)
• 1957년부터 시판
• 불면증, 감기 등에 진통/진정 효과:
“Wonder drug”
• 임신초기 입덧에 효과적인 항구토 작용
3
4.
탈리도마이드사건 후 미국의약물감시 변화
1962~ 1993~ 2007~ 2010~
• 영국 황색카드시스템 (1964~)
• 일본 유해사례보고(1967~)
• WHO-웁살라모니터링센터 (1968~)
4
시판후
유해사
례 보고
의무화
매드워
치 온라
인
보고실시
FDAAA
시판후
약물감
시
강화
유익성
위해성
평가
5개년
계획 발표
5.
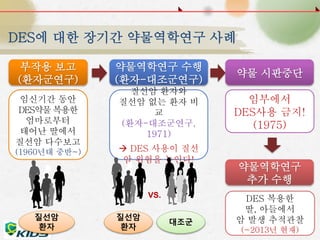
DES에 대한 장기간약물역학연구 사례
부작용 보고
(환자군연구)
약물역학연구 수행
(환자-대조군연구)
임신기간 동안
DES약물 복용한
엄마로부터
태어난 딸에서
질선암 다수보고
(1960년대 중반~)
질선암 환자와
질선암 없는 환자 비
교
(환자-대조군연구,
1971)
DES 사용이 질선
암 위험을 높인다!
질선암
환자
질선암
환자
대조군
VS.
약물 시판중단
임부에서
DES사용 금지!
(1975)
약물역학연구
추가 수행
DES 복용한
딸, 아들에서
암 발생 추적관찰
(~2013년 현재)
6.
Vioxx 시판철회와 안전관리체계변화
6
1999.5
• Vioxx 시판 시작 (Merck사, COX-2 inhibitor)
– 시판후 5년동안 전 세계적으로 8천만명 복용
– 전 세계적으로 연매출 USD 20 billion (2조) 이상
안전성 문제제기
(심혈관계 부작용)
2004.9 • Merck사, Vioxx 자진회수 결정
• 2004년 FDA CDER: 의약품 안전관리강화계획발표
• 2006년 Institute of Medicine (IOM) 보고서:
시판후 안전관리 강화 필요성 제기
• 2007년 FDA법 개정(FDAAA 2007)
미국 FDA
7.
FDAAA 2007
•Section 905: Active Postmarket Risk Identification
and Analysis
– 시판후 의약품 부작용을 파악하고 분석하기 위하여,
여러 대규모 자료원을 연계하여 분석하는 체계를 구축
한다.
• 2010년 6월 1일까지 최소 2,500만명
• 2012년 6월까지 최소 1억명의 데이터
– 공적 청구자료, 사보험 및 건강관련 전자기록을 포함함.
• 전자건강기록을 활용한 국가 의약품 안전성
평가 및 관리체계를 구축한다는 목표
• 이를 위하여, “Mini-Sentinel”이라는 파일럿
프로젝트를 시행 중임.
8.
8
의약품 부작용에따른 사회경제적 손실
미국에서는 정상적인 처방, 조제, 투약에도 불구하고
1994년 한 해 동안 약물 부작용으로 약 220만명
이상 입원하고 10만명 이상 사망한 것으로 추정
입원 및사망, 경제적 손실
약물 유해반응 으로 인한
미국 내 사망원인 4위
*미국의학원(Institue of Medicine, 1999)
9.
일본, 영국, 독일의의약품부작용 피해
• 일본 (인구 1.27억)
2001년 후생노동성에 보고된 부작용 사례 26,545건 중 사망 사례
1,239건. 일반의약품은 포함되지 않아 실제 사망자는 훨씬 많을 것이
라고 강조함.
• 영국 (인구 0.63억)
약물유해반응에 대한 코호트연구 138개에 대한 연구 결과 약물유해반
응으로 인한 입원이 전체 병원 입원기간의 약 4%에 해당하며, 영국
NHS 예산 중 약물 부작용에 대한 소요비용은 연간 £3.80억원 (약
6,500억원)에 이름
• 독일 (인구 0.81억)
2003년-2006년 기간 중 약물유해반응과 관련된 ICD-10코드로 진단
받은 환자에 대해 분석한 연구 결과, 전체 입원환자의 5%가 약물 유해
반응 관련 질환으로 입원하였고, 0.7%는 매우 관련성이 높은 것으로
나타났으며, 입원 중 약물 부작용을 경험한 환자는 4.5%였음.
9
10.
약물감시의 정의
WHOdefinition : Science and activities relating to the
detection, assessment, understanding and prevention of
adverse effects or any other drug-related problems
시판 약물의 의도되지 않은 효과 또는 실마리정보
의 지속적인 모니터링
약물의 유해작용 또는 약물관련 문제의 탐지, 평가,
해석, 예방에 관한 과학적 연구 및 활동
11.
시판전
안전성 정보
시판후
안전성 정보
11
허가시, 안전성 데이터의 한계
시판후, 새로운 안전성정보 파악
- 드물게 발생
- 장기복용으로 인한 발생
- 실제 진료사항 반영
- 약물오남용으로 인한 효과
의약품 시판후 안전관리
허가 취소, 시판 철회
12.
신약개발과 병행한 의약품안전관리체계 구
축
12
전임상실험
신약
개발 임상시험
허가신청
시판허가
제품 및
제품정보 향상
약물
감시
시판후
안전성
평가
허가취소,
시판철회
시판초기
안전성 정보 수집
사회 경제적
기여
약물부작용
최소화
시판전 파악된 효
과 및 안전성의 정
량적 평가가능
보다많은환자
상호작용
새로운
안전정보파악
실제 진료상황 반영
약물오남용파악
약물감시의 중요성
16.
시판후 약물감시의 궁극적목적
부작용 자
료수집
실마리정보 검색
약물역학연구
정책
반영
국민보건
향상
16
17.
Japan
(1967년)자발적
부작용보고 실시
(2002년) 보건의료인
의무보고 실시
피해구제 사업 실시
MIHARI 프로젝트
국외 의약품 안전관리 현황
WHO-UMC
(1968)국제약물감시센터.
WHO에서 운영
세계 국가로부터 약물 부작용 정보
축적 및 교류 활성화
‘14. 상반기 기준, 140여 개국의
930만 건 이상축적
USA
(1962~)탈리도마이드 사건
이후 자발적 시판후
유해사례 보고제도 도입
(1993~)메드워치 온라인
보고 실시
(2007~)FDAAA. 시판 후
약물감시 강화
(2010~)유익성/위해성 평가
5개년 계획 발표
Korea?
EU
[영국] 황색카드체계를 활용한
유해사례보고시스템 도입(1963)
[프랑스] 지역약물감시센터
운영(1973)을 통한 약물유해반응
수집 및 인과성 평가 실시
17
캐나다
연방보건부(HC).
치료제품의 안전성, 유효성
및 접근성을 증진시키기
위한 프로젝트 실시
약물감시프로그램 실시 (1988년) 자발적 부작용보고
실시
(2012년) KIDS 설립
(2014년) 피해구제제도 시행
18.
미국 - 시판후 안전성 모니터링
메드워치 프로그램 (MedWatch program) 운영 (FDA CDER)
보건의료인, 소비자 자발적 부작용 보고제도 + 제약회사 의무 보고
실마리정보 검색, 잠재적 실마리정보와 새로운 안전성 정보 분기별
웹페이지 공개
필요한 경우 제조회사 시판후 임상시험 (Phase IV trial) 실시 요구
기존 약물, 신약 대상 제약회사 위해평가 완화전략 (REMS) 수립
의학문헌, 국외 규제기관 정보 교류
실마리정보에 대한 인과성평가를 위해 대규모자료 연계 연구 수행
(Sentinel Initiative): Post-licensure Rapid Immunization
Safety Monitoring System (PRISM)
Mini-Sentinel 일부로 백신안전성 분야 연구 수행,
4개 national health plan 1억1천만 명의 의료기록, 약국조제기록,
실험실 검사 기록 및 방사선 검사 기록 이용,
9개 예방접종등록사업 및 10개 출생등록자료와 연계하여 연구 수행
(완료(2), 수행 중(5))
19.
미국 - 백신안전성 관리체계
Centers for Disease
Control and
Prevention (CDC)
National Center for
Emerging and Zoonotic
Infectious Diseases
(NCEZID)
Division of
Healthcare Quality
Promotion (DHQP)
Immunization
Safety Office
(ISO)
Health Resources
and Services Admin
(HRSA)
Food and Drug
Admin
(FDA)
National
Institutes
of Health
(NIH)
Dept of Health and
Human Services
(HHS)
National Vaccine
Program Office (NVPO)
Center for
Biologics
Evaluation and
Research
(CBER)
Office of
Biostatistics
and
Epidemiology
Silver Spring, MD Atlanta, GA
정책방향 결정, 기관간 협조
기초과학 및
임상연구
규제 및 집행 조사, 연구, 예방, 교육
국가백신피해구제
사업 관리
FDA/CDC 자발보고
시스템 공동 운영
Vaccine Adverse Event
Reporting System
(VAERS)
20.
영국 - YellowCard Scheme
MHRA와 Commission on Human
Medicines(CHM)이 함께 운영하는
자발적유해사례보고시스템(1963년
부터 실시)
2002년, Yellow Card Website가 개
설되어 전자보고시스템이 구축되었
으며, 2008년 추가 개발됨
영국은 Yellow Card Scheme을
통해 의약품과 백신 유해사례를
함께 수집하고 있음.
<영국의 Yellow Card Scheme의 역사적 흐름 도식화>
21.
영국 - YellowCard Scheme
Yellow Card Scheme 과정
연구결과 및 고찰
Provision of
information
Commit to data
base
QA of reports
received
Risk-benefit evalu
ation and
Assessment
Signal detection advice from CHM
Yellow Cards -
Adverse Drug
Reaction reports
Signal Evaluatio
n and
Prioritisation
Regulatory action
& communication
Acknowledgment
and/or follow-up
for more info
국내외 약물감시 관련홈페이지
국가 웹페이지 관련기관
한국 KIDS 의약품유해사례보고시스템(http://www.,drugsafe.or.kr) 식약처
한국의약품안전관리원
WHO Global Vaccine Safety (http://www.who.int/vaccine_safety/en/)
WHO-Uppsala Monitoring Center (http://www.who-umc.org/)
CIOMS Vaccine Pharmacovigilance
(http://www.cioms.ch/index.php/vaccine-pharmacovigilance)
WHO-UMC
(웁살라모니터링센터)
미국 FDA
(http://www.fda.gov/downloads/AboutFDA/CentersOffices/OrganizationC
harts/UCM291886.pdf)
CDC (http://www.cdc.gov/maso/mab_Charts_CIO.htm)
Vaccine Adverse Event Reporting System (https://vaers.hhs.gov/index)
FDA CDER
FDA CBER
CDC
영국 영국 MHRA (http://www.mhra.gov.uk/)
영국 Yellow Card Scheme (http://yellowcard.mhra.gov.uk/)
MHRA
네덜란드 네덜란드 국가약물감시센터 Lareb (http://www.lareb.nl) Lareb
캐나다 캐나다 유해사례 보고시스템 (CAEFISS) (http://www.phac-aspc.gc.ca/im/vs-sv/
index-eng.php)
호주 호주 유해사례시스템 (Australian Adverse Drug Reactions System, ADRS)
(http://www.tga.gov.au/safety/daen.htm)
TGA
일본 일본 PMDA (http://www.pmda.go.jp/english/) PMDA
중국 중국 CFDA (http://eng.sfda.gov.cn/WS03/CL0755/) CFDA
24.
국내 의약품 안전관리의발전과정
• 의약품등 재평가제도 (1977~)
• 자발적부작용보고제도 (1988~)
• 의약품 피해구제제도 도입 (1991)
• WHO-UMC의 회원국가로 등록 (1992)
• 신약 등의 재심사제도 (1995~)
• 제약회사 및 약국 : 15일 이내 의무보고 신설 (2004~)
• 제약회사 안전관리책임자 지정 의무화 (2007~)
• APEC PV분야 챔피언 국가 선정(2011.9)
• 한국의약품안전관리원 설립 (2012.4.17)
• 자발적부작용보고자료 실마리평가알고리즘개발(2012.7)
• 의약품유해사례보고시스템(KAERS)으로 유해사례 접수 (2012.10.1)
• 지역의약품안전센터 27개 지정 및 운영(2014.1.1)
• 국외 발생 유해사례보고 의무화(2014. 8~)
• 안전관리책임자 교육기관 지정 및 교육의무화(2014.9~)
• 의약품 피해구제제도시행 (2014.12~)
24
의약품 등 피해구제제도 대상
의약품을 적정하게 사용한 사람이 의약품의
부작용으로 인하여
> 질병(입원을 요하거나 이와 동등한 정도)에 걸리거나
> 장애가 발생하거나
> 사망한 경우
27
28.
포괄적 안전성 정보수집·분석 기반 마련
국내외 시판 후 안전성정보 관리
국내 발생 유해사례 국외 발생 유해사례
WIRTE THE TEXT HERE WIRTE THE TEXT HERE 국외 안전성 정보 수집 관련
규정 개정 및 시행(2014.8~)
국제표준에 맞는 유해사례 용
어/보고형식 도입
유해사례보고건수 지속 증가
및 보고 충실도 제고
소비자 보고 활성화
안전관리책임자 및 실무자 역
량 강화(의료기관/제약회사)
28
29.
국내 유해사례보고 DB구축및 관리 절차
부작용
보고자료
접수
DB질관리
(스크리닝/
충실도 점검)
DB 구축
(클렌징)
실마리정보
검색
데이터마이닝
기법을 이용한
지표 산출
안전성정보
분석·평가
안전성정보
제공
(허가사항
변경안,
보고서 등)
29
국내외
보고자료
교류
30.
• 접수 및처리
– 온라인 보고 → 홈페이지의 유해사례보고시스템(KAERS)을 통하여 입력
– 오프라인 보고 : 이메일, 팩스 및 우편 접수 → KAERS 입력
– 전화 접수 : 의약품부작용신고센터 전화 상담 → KAERS 입력
접 수
[오프라인]
E-mail,
Fax,우편
KAERS
[온라인]
대표포털,
내부포털
[전화]
부작용
신고센터
직접 입력
처 리
유해사례보고시스템
부작용신고센터
유해사례 접수 경로
30
31.
의약품유해사례보고시스템 운영(2012.10.1~)
KIDSWebsite (http://www.drugsafe.or.kr)
유해사례 보고
Korea
Adverse
Event
Reporting
System
유해사례 보고자
• 소비자용
• 의약전문가용
• 제약회사용
31
의약품유해사례보고서 서식 개발
간편서식 기본서식
양식
사용자 구분 일반 소비자
제조수입업체
지역의약품안전센터
의약전문가 등
특징
일반인의 보고 편의성 고려
직관성 고려한 디자인
국제표준 필수항목 반영
다양한 사용자 그룹 고려
필수 입력항목
(공통)
환자정보, 의심되는 의약품명, 유해사례명, 보고자정보
33
34.
약사법 제68조의 8(부작용등의 보고)
제68조의8(부작용 등의 보고) ① 의약품등의 제조업자·품
목허가를 받은 자·수입자 및 의약품 도매상은 의약품 등
으로 인하여 발생하였다고 의심되는 유해사례로서 질병·
장애·사망, 그 밖에 보건복지부령으로 정하는 의약품등의
안전성·유효성에 관한 사례를 알게 된 경우에는 의약품안
전관리원장에게 보고하여야 한다.
② 약국개설자와 의료기관개설자는 유해사례를 의약품안
전관리원장에게 보고하여야 한다.
34
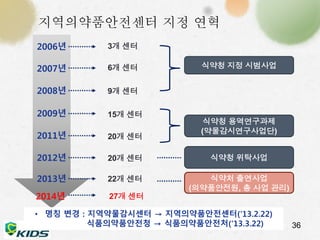
지역의약품안전센터 지정 연혁
3개 센터
6개 센터
9개 센터
15개 센터
20개 센터
20개 센터
식약청 지정 시범사업
식약청 용역연구과제
(약물감시연구사업단)
식약청 위탁사업
22개 센터 식약처 출연사업
(의약품안전원, 총 사업 관리)
• 명칭 변경 : 지역약물감시센터 → 지역의약품안전센터(‘13.2.22)
식품의약품안전청 → 식품의약품안전처(‘13.3.22)
2006년
2007년
2008년
2009년
2011년
2012년
2013년
36
2014년 27개 센터
2014 지역센터 구성도
센터
#1
센터
#2
센터
#25
센터
#26
센터
#27
25개 대학병원 국립중앙
의료원
대한약사회
약국
보건소 등
지역 약국∙의원 및 2차병원 공공의료조직
포괄적인 약물감시 안전망 구축
38
39.
지역센터 운영체계
지역의약품안전센터
(전용사무실 및 전담인력 구비)
의원
약국
학회 회원
공공
의료조직
약물유해사례
모니터링
집중모니터링
약물유해사례
상담
교육 및 홍보
- 원내 모니터링
- 관할 지역 약물
유해사례 수집
- 인과관계 검토
- 수집자료 보고
- 취약계층
(노인/소아)
- 식약청 지정
의약품
- 자료수집 및
인과관계 검토
- 전담인력 상주
- 소비자 약물
유해사례 상담
- 보고자 애로
센터 운영
- 센터 내부인력 :
지역센터외
신규 및 보수
교육
- 의약사 연수
교육
- 의약대생 정규
교육
39
40.
40
2013년 22개지역센터 지정
Total : 567,295
2014년 27개 지역센터 지정
2011년 20개 지역센터 지정
2009년 15개 지역센터 지정
6,239
2006년 3개 지역센터 지정
13 21 14 21 57 76 40 13 19 378 290 264 117 158 199 620 1,400
27,010
14,453 12,796
74,657
64,143
183,260
92,375
200,000
180,000
160,000
140,000
120,000
100,000
80,000
60,000
40,000
20,000
0
No. of AEs reported
Year
88,680
국내 의약품 유해사례 접수현황
41.
41
WHO-UMC 유해사례보고순위(인구 백만명당)
출처: Uppsala reports 65 (2014.4)
42.
2013.8. 한국 8위2014.2. 한국 5위
42
WHO-UMC 유해사례 보고순위(절대 보고건수)
1.미국
2.영국
3.독일
4.한국
2014.8. 한국 4위 5.캐나다
43.
자발적부작용보고자료의 활용
국제
교류
안전
조치
정보
전파
교육
정보
제공
• WHO –
UMC보고
• 코이카
의약품규
제연수교
육실시
한국이 세계 2위
44.
안전성 평가 우선순위약물 선정
1. 안전성 이슈 제기
2. 다빈도 보고 (자발적 부작용 보고 자료)
3. 국외 실마리정보 탐지 (FDA, WHO 등 국외에서
제기된 안전성 속보 등
4. 다빈도 처방(건강보험청구자료)
5. 신약
한국의약품안전관리원 “안전성평가위원회” 자문
실마리정보 검색을 위한 약물 우선순위 선정
44
KAERS 원시자료 DB구축
KAERS DB
기본(환자)정보
DB
의약품정보
DB
유해사례정보
DB
중대사례정보
DB
보고자정보
DB
평가정보
DB
48.
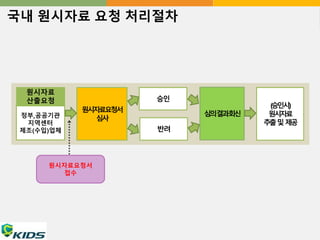
국내 원시자료 요청처리절차
원시자료
산출요청 승인
원시자료요청서
심사
심의결과회신
(승인시)
원시자료
추출및 제공
정부,공공기관
지역센터
제조(수입)업체
원시자료요청서
접수
반려
49.
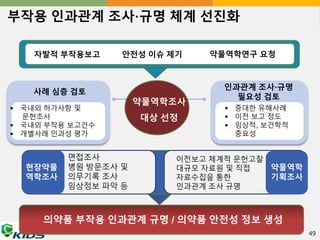
부작용 인과관계 조사·규명체계 선진화
자발적 부작용보고 안전성 이슈 제기 약물역학연구 요청
약물역학조사
대상 선정
인과관계 조사·규명
필요성 검토
중대한 유해사례
이전 보고 정도
임상적, 보건학적
중요성
사례 심층 검토
국내외 허가사항 및
문헌조사
국내외 부작용 보고건수
개별사례 인과성 평가
현장약물
역학조사
약물역학
기획조사
면접조사
병원 방문조사 및
의무기록 조사
임상정보 파악 등
이전보고 체계적 문헌고찰
대규모 자료원 및 직접
자료수집을 통한
인과관계 조사 규명
의약품 부작용 인과관계 규명 / 의약품 안전성 정보 생성
49
인식 개선을 위한대국민 교육·홍보(1)
[KTX서울역 열차 내 광고,
2013.9]
[부작용신고 CC공모전, 2014.3]
“부작용신고 활성화 공익캠페
인”“ 의약품을 복용한 후 부작용이
생기면 꼭 신고해 주십시오.
여러분의 소중한 신고로
내 가족과 이웃의 부작용 피해를
[TBS교통방송, 줄일 수 있습니다.”
2014.7.1~9.30] 51
52.
인식 개선을 위한대국민 교육·홍보(2)
[의약품 안전사용 교육자료] [HPV예방백신 안전사용 안내]
[어린이 의약품 안전사용 안내] [임신부 의약품 안전사용 안내] 52
53.
어린이에 대한 안전사용을위한 주의사항
어린이는 어른의 축소판이 아닙니
다!
어린이는 간의 대사기능, 신장의 배설기능이 어른과 같
지 않으므로 같은 용량의 약물에서도 효과와 부작용이 달
라질 수 있으므로, 임의로 복용하지 않습니다.
어린이용 의약품이라도 마음대로 먹으면 부작용(두드러
기, 설사 등)이 나타날 수 있어요.
약 복용 시, 약 이름을 잘 기억해 두세요.
54.
어린이를 위한 의약품사용안내
1. 약과 아이 이름 보고
약 이름, 아이 이름을 꼭 확인
약의 종류와 용량은 아이의 질병상
태와 체중에 따라 달라요.
친구나 형제, 자매끼리 약을 나누어
먹이지 마세요.
2. 정확한 용량 보고
계량컵∙스푼∙의약품주입기 사용
어른을 기준으로 어린이 용량을 유
추하여 먹이지 마세요.
약을 먹을 시간과 용량을 적어두는
습관을 기르세요.
55.
어린이를 위한 의약품사용안내
3. 유통기한 보고
유통기한 지나면
과감히 버리기
특히, 물약은 오래두면
변질되기 쉬워요.
환경을 위해 약국의 폐
의약품 수거함에 버려
주세요.
4. 보관장소 보고
어린이 손에 닿지
않는 곳에 보관하기
어린이의 손에 닿는 곳
에 보관하면 우발적인
중독사고의 원인이 됩
니다.
5. 부작용 보고
부작용이 생기면
신고하세요
한국의약품안전관리원
으로 신고하세요.
응급상황일 때에는 응
급실 또는 119구급대로
전화하세요.
56.
임산부에서 안전사용을 위한주의사항
원칙적으로 임신기간 동안 약을 임의로 복용하지 않는
것이 좋으며, 약물로 인하여 태아에게 기형을 유발 할
수 있는 약물은 복용하지 말아야 합니다.
약물의 효과로 인한 유익성이 위험을 상회할 경우, 전
문가의 판단 및 감독하에,
약물을 복용할 수 있습니다.
약물복용 시에는 항상 의사 또는 약사와
상담하여야 합니다.
57.
임신 중 의약품사용해도 되나요?
☆ 임신시기별 약물이 태아에 미치는 영향 ☆
착상
전기
기관
형성기
태아기
약물 노출로 유산이 될 수 있으나, 유산될 정도가 아니라면 완전히
회복되어 정상적으로 성장할 수도 있습니다.
가장 민감한 시기로 약물 사용을 최소화해야 합니다.
태아의 외부 생식기가 형성되고 발달하는 시기로 성호르몬에 영향
을 줄 수 있는 약물 노출에 주의해야 합니다.
58.
임신 중 의약품사용해도 되나요?
☆ 임부금기 1등급☆
태아에 대한 위해성이 명확해 원칙적으로 임신 중에 사용 금기인 약물
59.
인유두종바이러스(HPV) 예방백신
백신접종효과는
자궁경부암, 자궁경부 상피 내 종양 등의 질병을 예방할 수 있습니다.
백신접종 대상은
인유두종바이러스(HPV)에 의한 자궁경부암 예방을 원하는
9~26세 여성이 주요 대상입니다.
백신접종 전에는
백신 접종이 가능한 건강상태인지, 효과와 부작용은 무엇인지
의사와 상의하신 후 접종 여부를 결정하세요.
백신접종 후에는
약 30분간 접종한 의료기관에서 휴식을 취하면서
유해사례(이상반응) 발생여부를 관찰하셔야 합니다.
조영제 안전관리 길라잡이리플릿(2014.9)
61
• 지역센터-의약품안전원 공동 발간
• 소비자 및 의약전문가를 대상으로 조영제 사용시 안전관리 사항을 안내
62.
항암제 부작용 대처하기리플릿(2014.9)
• 지역센터-의약품안전원 공동 발간
• 소비자를 대상으로 항암제 투여 후 발생될 수 있는 부작용에 대한 대처방법을 안내
62
63.
포괄적인 의약품안전체계 구축
지역의약품안전센터 협회단체 환자 및 보호자
의사,약사, 제약업체
간호사 등
한국의약품
안전관리원
DB구축
및 연계
의약품
안전성
근거평가
교류
정보수집
건강보험자료
지역의약품안전센터 및
관련 학계 자료
병원 EMR,능동적
부작용 수집자료
통계청
한국암등록자료
행정안전부
제약업계
부작용보
고, 정보수
WHO-UMC, AsPEN,
외국정부, 국외언론
및 포털, 외교통상부
안전성정보 교환 집
정보보고
및 교류
위해교류 및 위해관리
부작용
보고DB
한국보건의료연구원
식품의약품
안전처
부작용보
고
WHO-UMC
64.
환자(보호자) 및 의약전문가
풍부한
자료 확보
약물의
안전한
사용
부작용
피해 예방
약물
오남용
방지
64
약물 안전망 구축으로 국민 보건 향상
유해사례(부작용)보고
















![Japan
(1967년) 자발적
부작용보고 실시
(2002년) 보건의료인
의무보고 실시
피해구제 사업 실시
MIHARI 프로젝트
국외 의약품 안전관리 현황
WHO-UMC
(1968)국제약물감시센터.
WHO에서 운영
세계 국가로부터 약물 부작용 정보
축적 및 교류 활성화
‘14. 상반기 기준, 140여 개국의
930만 건 이상축적
USA
(1962~)탈리도마이드 사건
이후 자발적 시판후
유해사례 보고제도 도입
(1993~)메드워치 온라인
보고 실시
(2007~)FDAAA. 시판 후
약물감시 강화
(2010~)유익성/위해성 평가
5개년 계획 발표
Korea?
EU
[영국] 황색카드체계를 활용한
유해사례보고시스템 도입(1963)
[프랑스] 지역약물감시센터
운영(1973)을 통한 약물유해반응
수집 및 인과성 평가 실시
17
캐나다
연방보건부(HC).
치료제품의 안전성, 유효성
및 접근성을 증진시키기
위한 프로젝트 실시
약물감시프로그램 실시 (1988년) 자발적 부작용보고
실시
(2012년) KIDS 설립
(2014년) 피해구제제도 시행](https://image.slidesharecdn.com/2014-141112183334-conversion-gate01/85/slide-17-320.jpg)












![• 접수 및 처리
– 온라인 보고 → 홈페이지의 유해사례보고시스템(KAERS)을 통하여 입력
– 오프라인 보고 : 이메일, 팩스 및 우편 접수 → KAERS 입력
– 전화 접수 : 의약품부작용신고센터 전화 상담 → KAERS 입력
접 수
[오프라인]
E-mail,
Fax,우편
KAERS
[온라인]
대표포털,
내부포털
[전화]
부작용
신고센터
직접 입력
처 리
유해사례보고시스템
부작용신고센터
유해사례 접수 경로
30](https://image.slidesharecdn.com/2014-141112183334-conversion-gate01/85/slide-30-320.jpg)

















![인식 개선을 위한 대국민 교육·홍보(1)
[KTX서울역 열차 내 광고,
2013.9]
[부작용신고 CC공모전, 2014.3]
“부작용신고 활성화 공익캠페
인”“ 의약품을 복용한 후 부작용이
생기면 꼭 신고해 주십시오.
여러분의 소중한 신고로
내 가족과 이웃의 부작용 피해를
[TBS교통방송, 줄일 수 있습니다.”
2014.7.1~9.30] 51](https://image.slidesharecdn.com/2014-141112183334-conversion-gate01/85/slide-51-320.jpg)
![인식 개선을 위한 대국민 교육·홍보(2)
[의약품 안전사용 교육자료] [HPV예방백신 안전사용 안내]
[어린이 의약품 안전사용 안내] [임신부 의약품 안전사용 안내] 52](https://image.slidesharecdn.com/2014-141112183334-conversion-gate01/85/slide-52-320.jpg)





























![New Drug Application [NDA]](https://cdn.slidesharecdn.com/ss_thumbnails/newdrugapplicationnda-160619063242-thumbnail.jpg?width=640&height=640&fit=bounds)

























![바이오 데이터 활용의 달인되기 : 데이터 3법 3시간 완성 세미나 [4강 이시항 변호사] 201006](https://cdn.slidesharecdn.com/ss_thumbnails/session4-201008081218-thumbnail.jpg?width=640&height=640&fit=bounds)





![[2013] 외국계 제약영업 노동자들의 직무스트레스 및 노동강도를 통해 살펴본 노동조건 개선방안 연구](https://cdn.slidesharecdn.com/ss_thumbnails/2013-140127195551-phpapp01-thumbnail.jpg?width=640&height=640&fit=bounds)




























