Downloaded 16 times
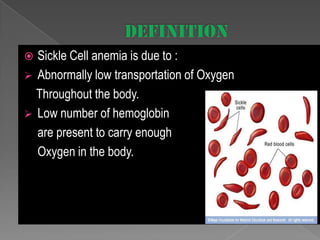




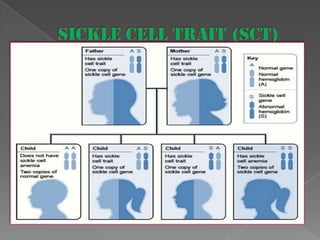

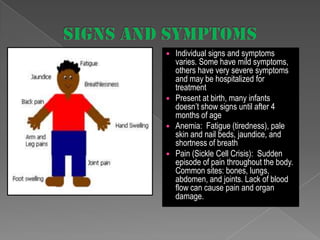
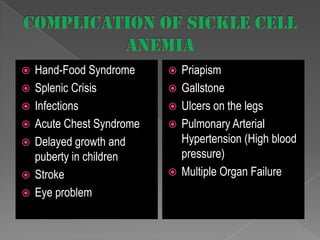










Sickle cell anemia is a genetic blood disorder caused by a mutation in the beta-globin gene. This mutation causes red blood cells to become rigid and sickle-shaped, which can block blood vessels and cause pain. It results in low oxygen transport and anemia. Symptoms vary but can include fatigue, pain crises, infections, and organ damage. Treatment focuses on prevention of complications, pain management, antibiotics, hydration, folic acid supplements, and transfusions in severe cases. While there is no cure, management has improved life expectancy significantly in recent decades.









































































