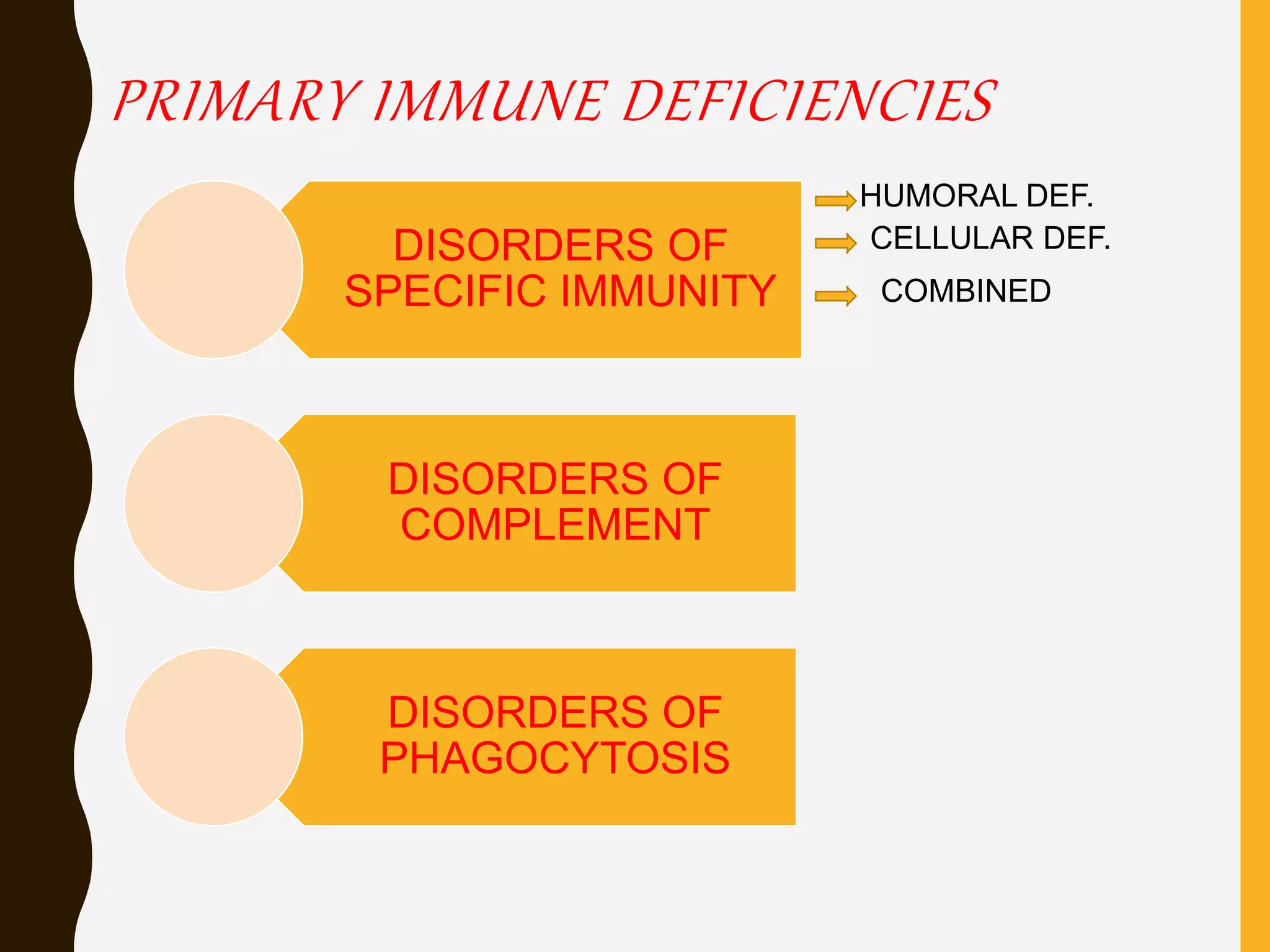
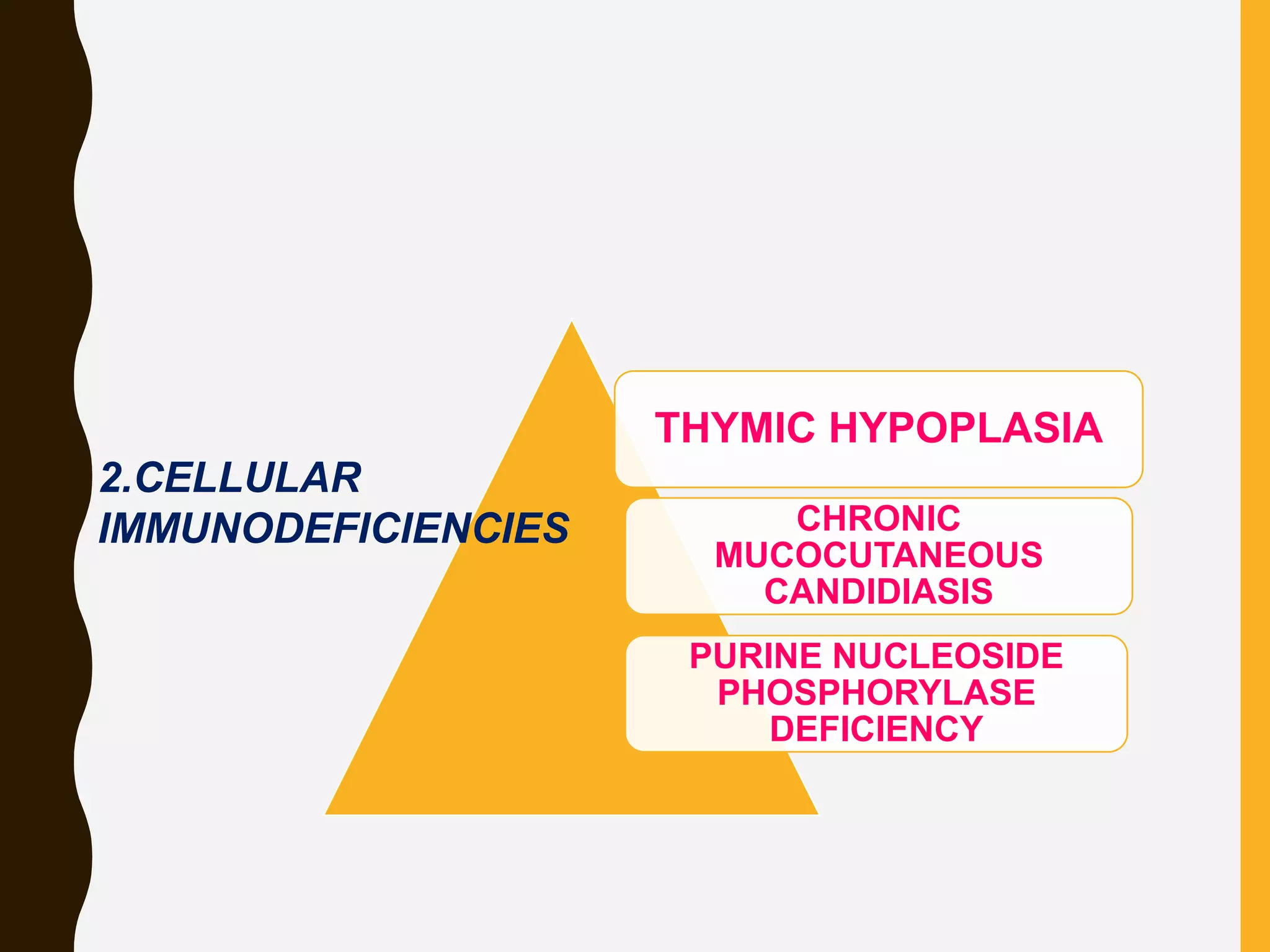
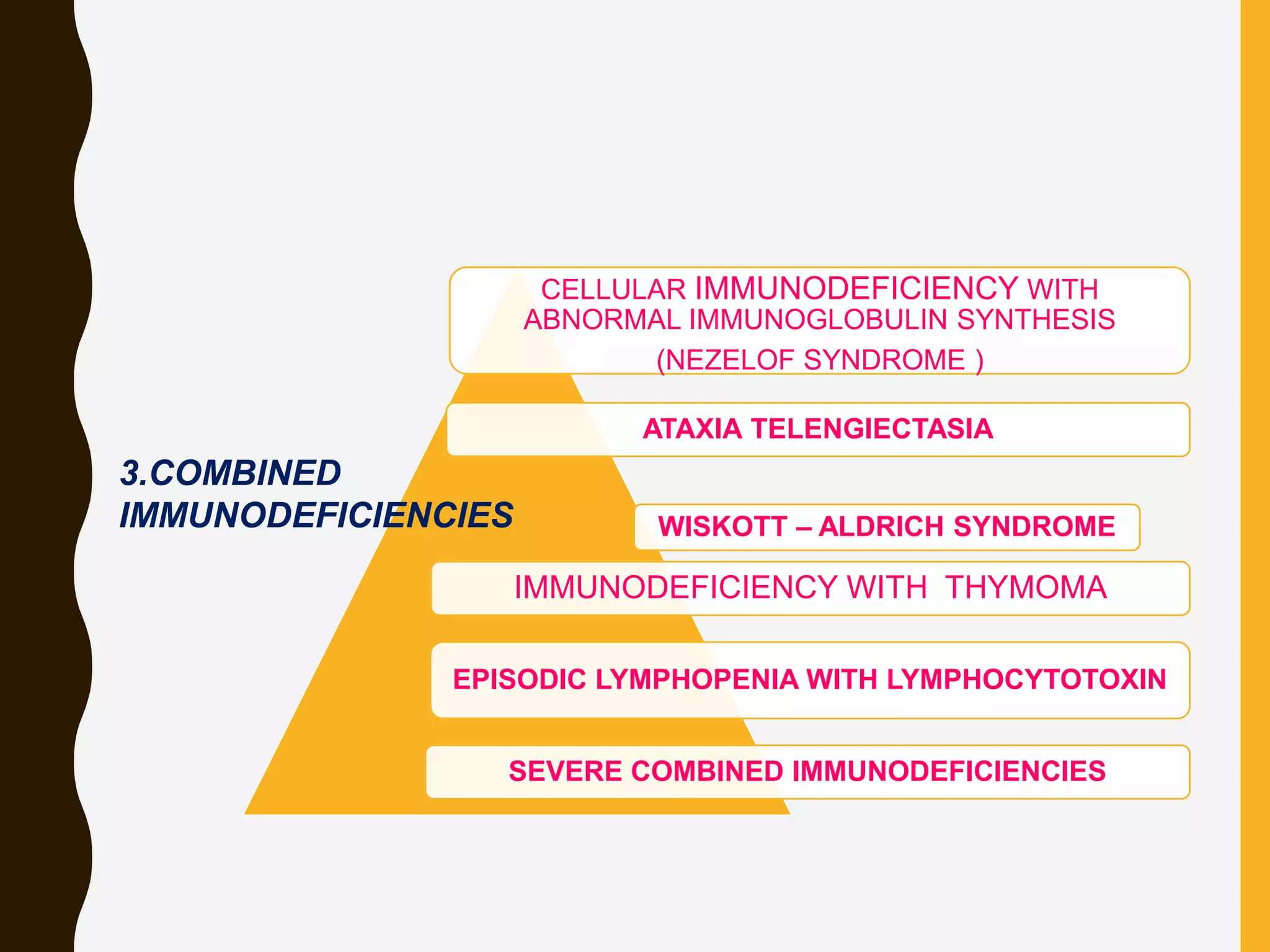
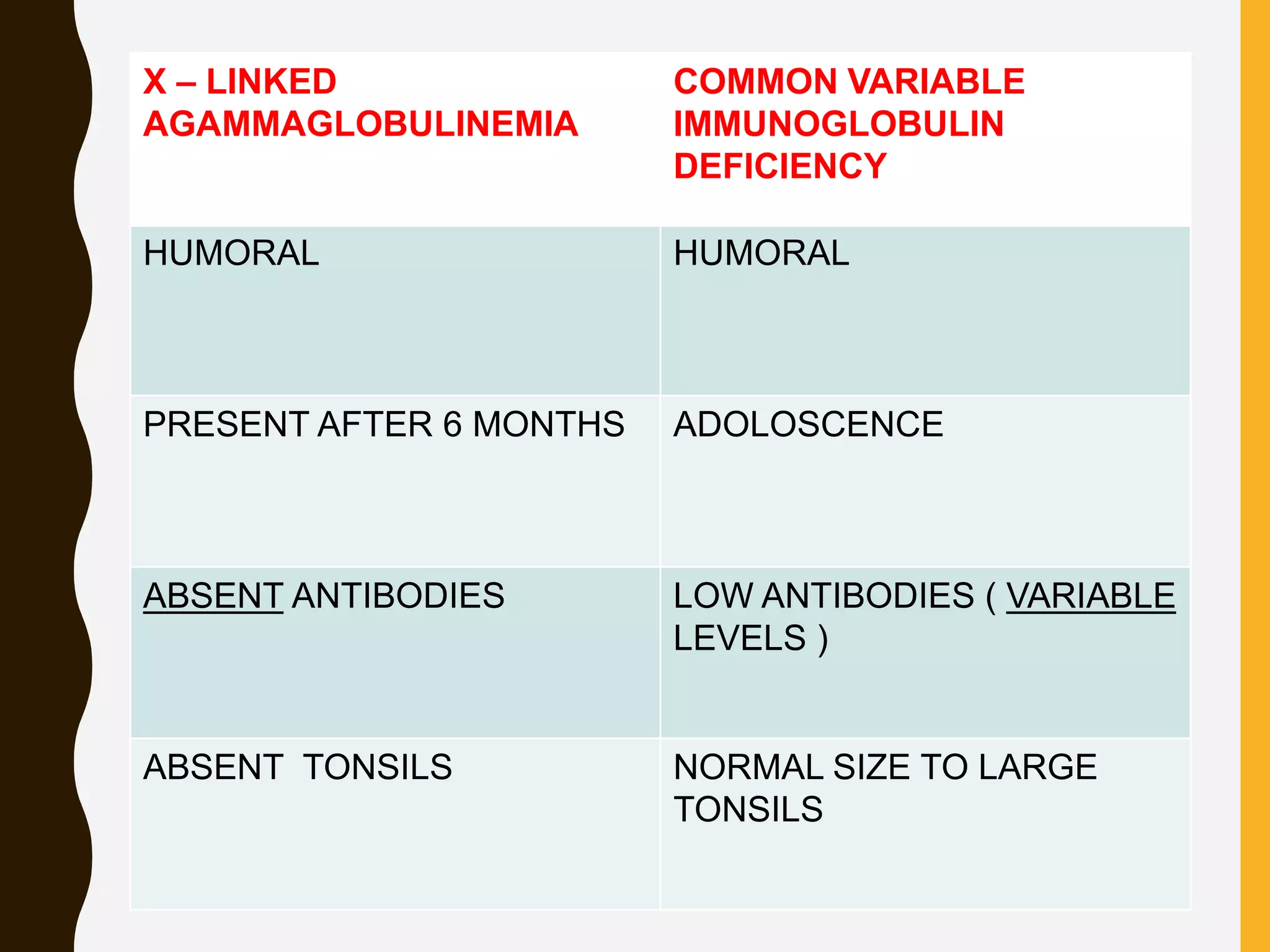
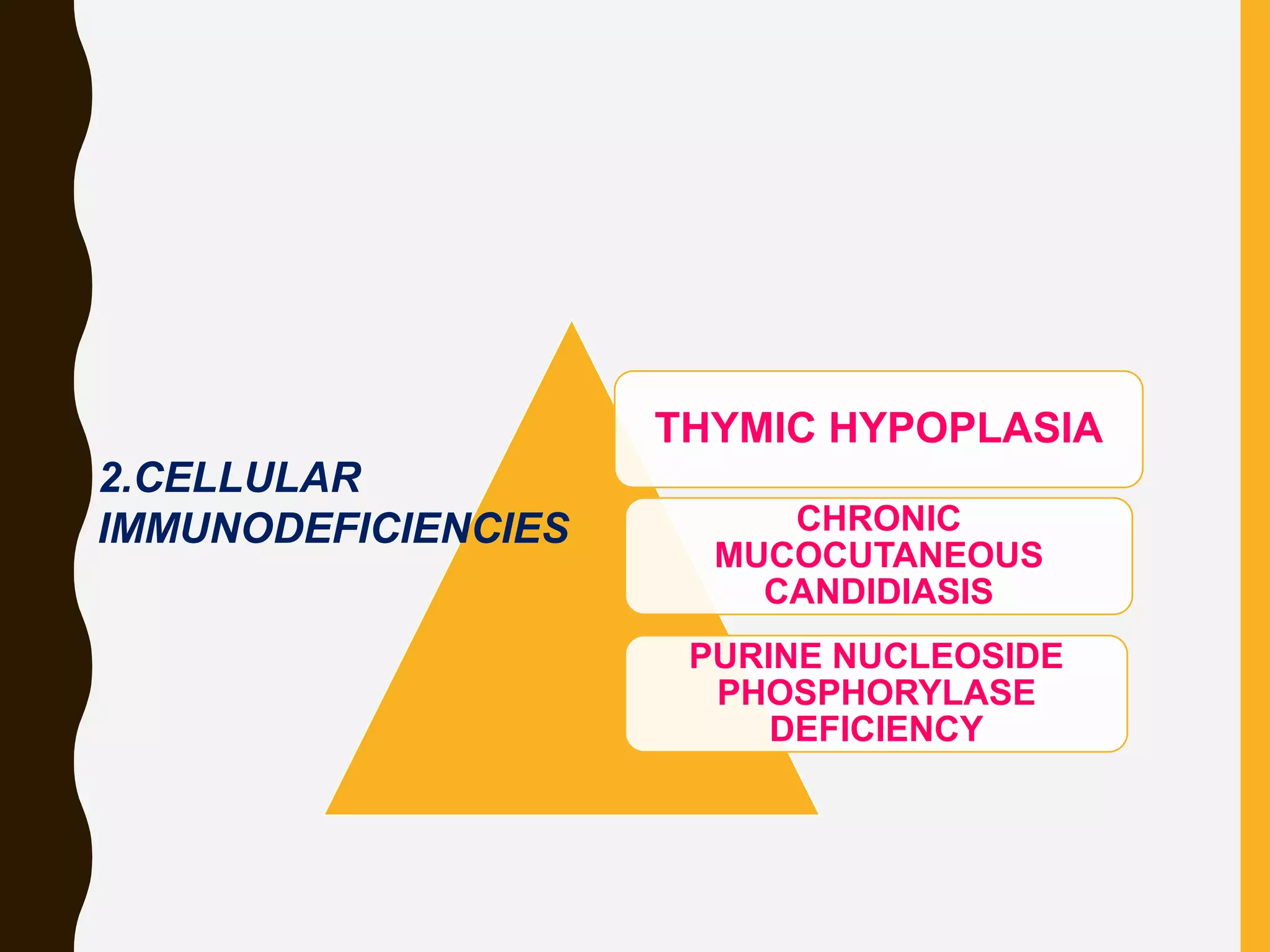
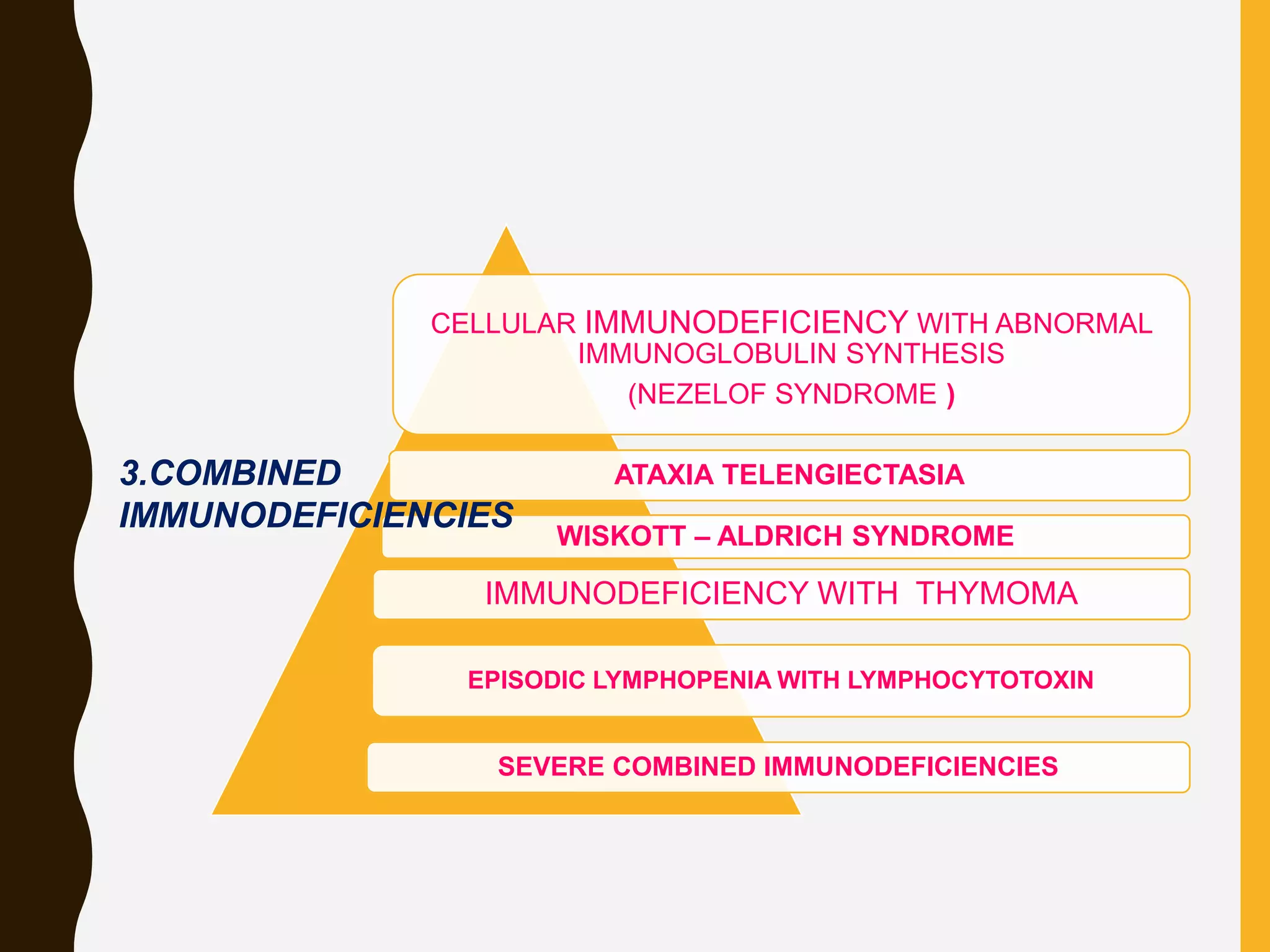
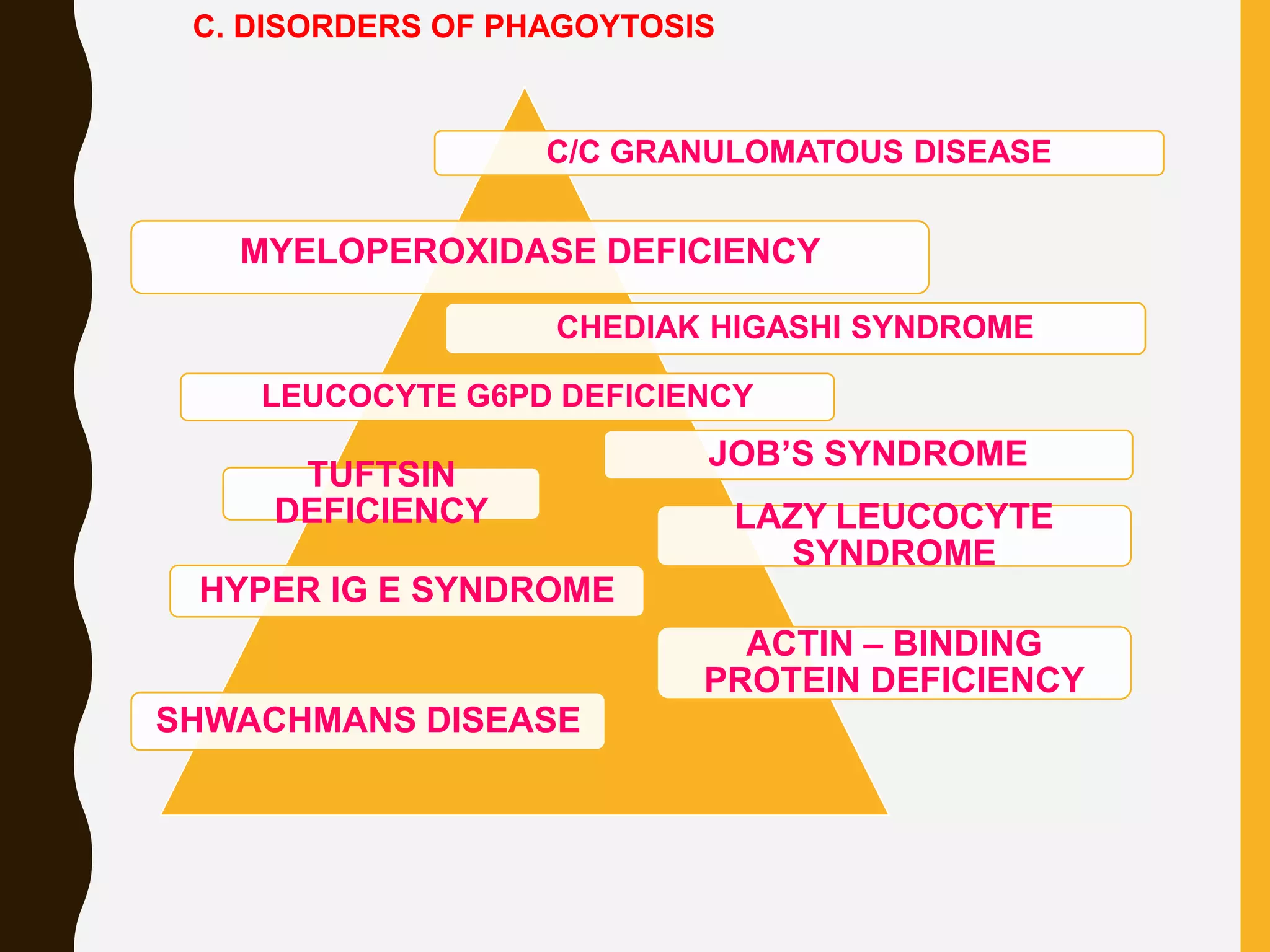
Primary immunodeficiencies are caused by genetic defects and present early in life, while secondary immunodeficiencies are acquired through diseases, medications, malnutrition etc. and are more common. The document describes various primary immunodeficiencies including X-linked agammaglobulinemia presenting in boys after 6 months with absent antibodies, and common variable immunodeficiency presenting in adolescence with low antibodies and recurrent infections. It also discusses secondary immunodeficiencies like AIDS which is caused by HIV infection of T cells leading to severe deficiencies.