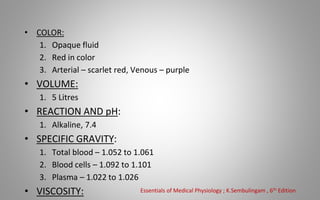
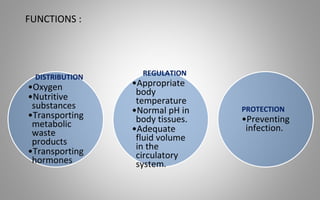
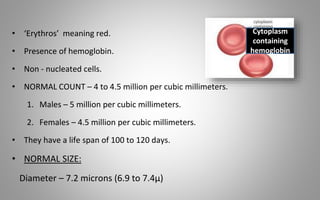
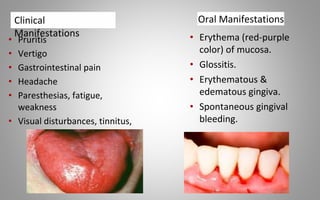
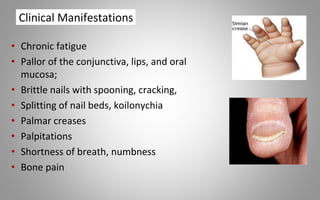
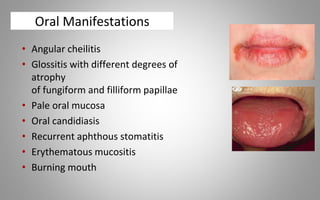
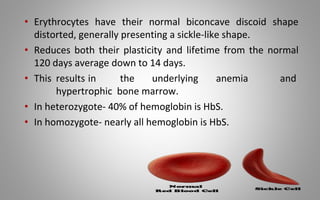
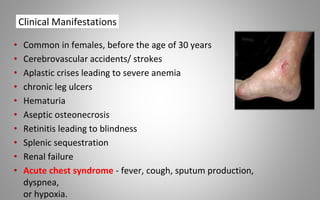
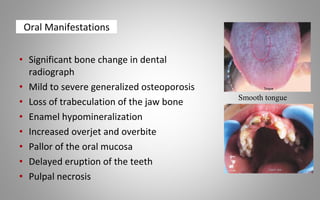
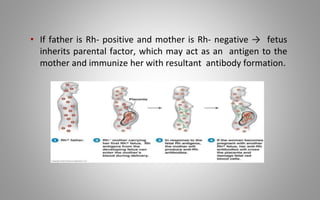
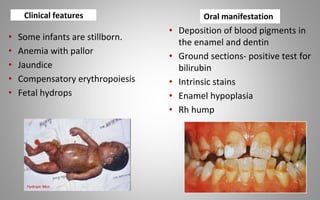
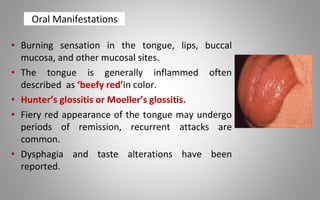
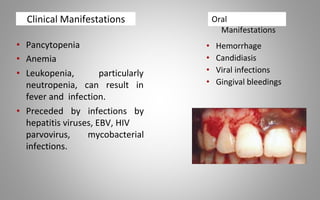
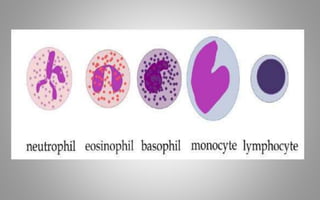
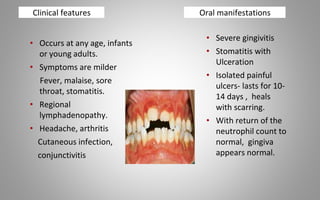
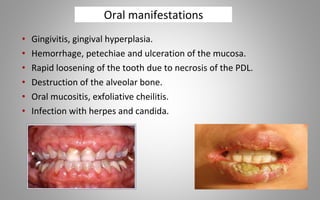
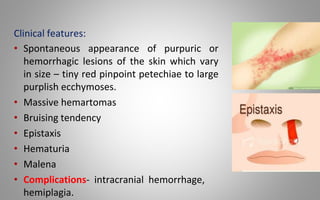
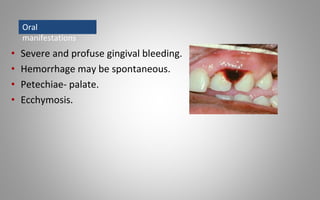
This document provides an overview of blood components, disorders, and implications for prosthodontics. It begins with an introduction to blood and its composition, functions, and components including red blood cells, white blood cells, platelets, and plasma. The document then discusses specific red blood cell disorders like anemias, sickle cell disease, and thalassemias. White blood cell disorders like leukopenia and leukemia are also summarized. Finally, the implications of blood disorders like anemia are discussed for prosthodontic procedures and treatments.