


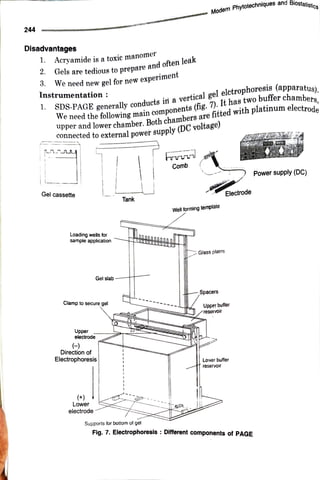




The document elaborates on the process of sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), a technique used to separate proteins based on their molecular weight. It discusses components necessary for gel preparation, the functionality of SDS in protein denaturation, and the setup of electrophoresis apparatus. Additionally, it outlines the advantages and disadvantages of polyacrylamide gels and provides protocols for gel assembly, sample preparation, and protein staining.























































































