Downloaded 89 times



![Reaction Kinetics
The study of rates of reactions is called kinetics
The order of a reaction is sum of the exponents of the
concentrations in the rate law – the first example is first
order, the second one second order.
NaOH + C
CH3
CH3
CH3 Br NaBr + C
CH3
CH3
CH3 OH
v = k[C4H9Br]
NaOH + NaBr +
v = k[CH3Br][NaOH]
CH3Br CH3OH](https://image.slidesharecdn.com/reaksisn-1sn-2e-1dane-2-130503030119-phpapp02/85/Reaksi-sn-1-sn-2-e-1-dan-e-2-4-320.jpg)










![SN1 Energy Diagram
Rate-determining step is
formation of carbocation
Step through highest energy
point is rate-limiting (k1 in
forward direction)k1 k2
k-1
V = k[RX]](https://image.slidesharecdn.com/reaksisn-1sn-2e-1dane-2-130503030119-phpapp02/85/Reaksi-sn-1-sn-2-e-1-dan-e-2-18-320.jpg)

























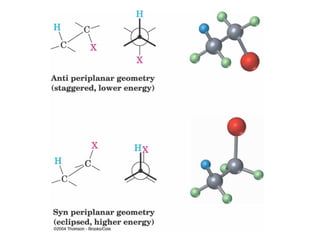



![11.14 The E1 Reaction
Competes with SN1 and E2 at 3° centers
V = k [RX]](https://image.slidesharecdn.com/reaksisn-1sn-2e-1dane-2-130503030119-phpapp02/85/Reaksi-sn-1-sn-2-e-1-dan-e-2-52-320.jpg)




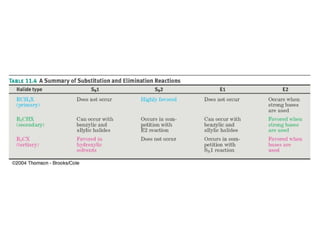


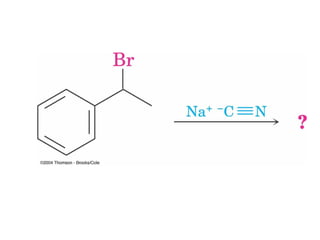
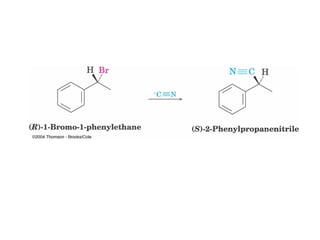




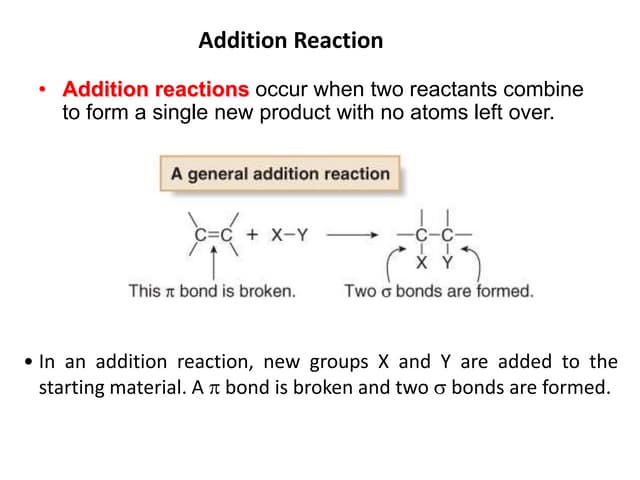
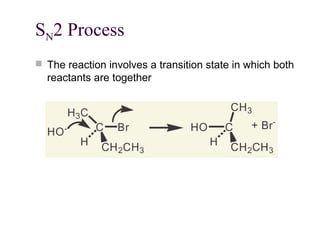
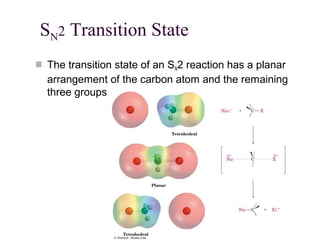
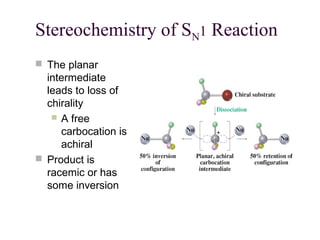
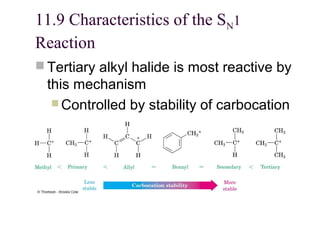
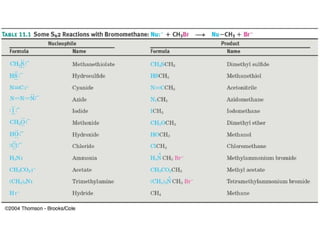
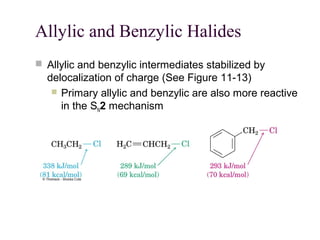
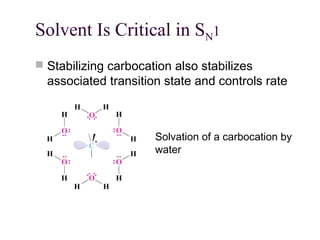
The document summarizes key concepts about nucleophilic substitution and elimination reactions of alkyl halides. It discusses the SN1 and SN2 substitution mechanisms, and the E1 and E2 elimination mechanisms. The SN1 reaction involves formation of a carbocation intermediate and proceeds through two steps, while the SN2 reaction occurs in a single concerted step with the nucleophile and leaving group interacting simultaneously. The E1 reaction eliminates the leaving group first to form a carbocation, while the E2 reaction involves a concerted elimination. Stereochemistry and factors affecting reactivity such as stability of carbocations, solvent effects, and steric hindrance are also covered.