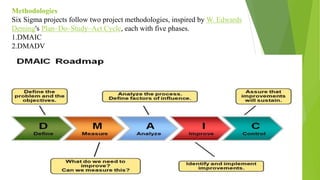
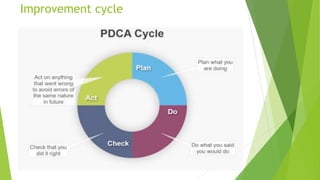
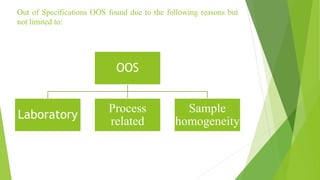
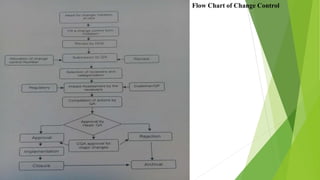
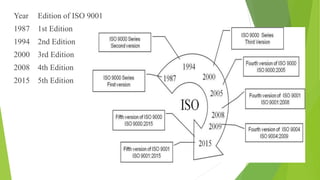
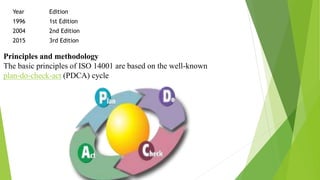
The document details quality management concepts including total quality management (TQM), quality by design (QBD), and six sigma, providing principles and methodologies to enhance product quality and operational efficiency. It emphasizes the importance of compliance with standards such as ISO 9000 and ISO 14000 for effective management systems and change control processes. Additionally, it outlines the significance of addressing out-of-specification results and conducting thorough investigations to ensure product reliability and safety.