












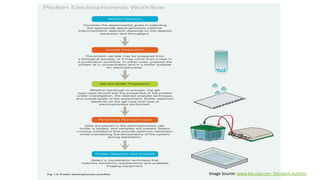
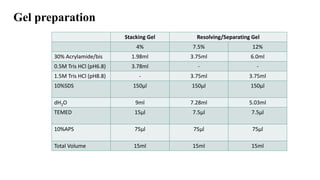
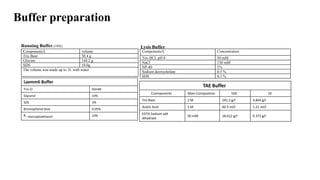

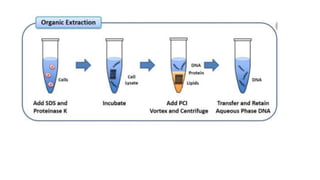





Sample preparation involves collecting and processing samples to isolate proteins, DNA, or other molecules of interest. For protein samples, cells or tissues are lysed to release proteins, which are then separated from other components through techniques like centrifugation or filtration. Purification methods separate protein mixtures based on properties like size, charge, or solubility using chromatography, electrophoresis, or precipitation. DNA extraction from blood involves lysing cells, precipitating DNA, and purifying it from other cell components. Proper sample preparation is essential for downstream diagnostic and analytical tests.

























































![Human Reproduction [ Reproductive System ] Notes @irfanullah_mehar Irfanullah...](https://cdn.slidesharecdn.com/ss_thumbnails/humanreproductionreproductivesystemnotesirfanullahmeharirfanullahmeharjanantantra-260111172350-56e85778-thumbnail.jpg?width=640&height=640&fit=bounds)









