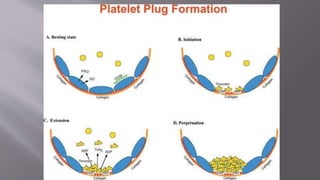
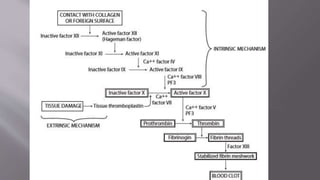
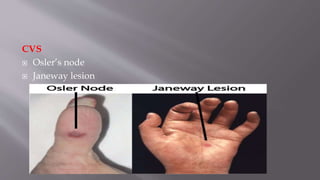
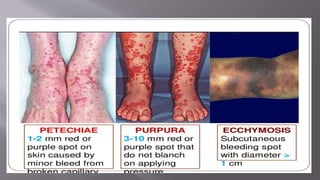
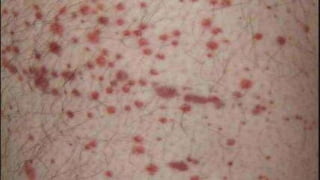
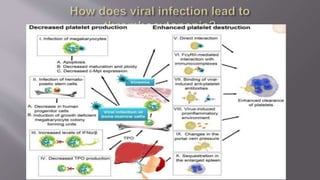
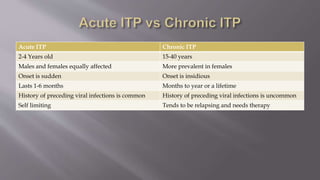
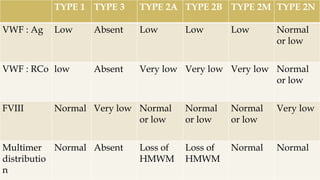
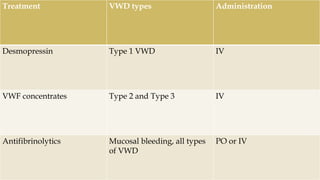
The seminar presentation covered hemostasis and approaches to bleeding disorders in pediatrics. It discussed the pathophysiology, clinical features, laboratory findings and management of idiopathic thrombocytopenic purpura, Von Willebrand's disease, and hemophilia. It provided an overview of hemostasis and the coagulation cascade, approaches to evaluating a child with bleeding, and specifics on selected bleeding disorders. The presentation included descriptions of laboratory tests used to evaluate coagulation factors and identify bleeding disorders.