

![2-qubit system: Molecular Magnet [Mn12(Rdea)]
contains two weakly coupled subsystems
5
M=Methyl diethanolamine M=allyl diethanolamine
Subsystem spin should not be identical](https://image.slidesharecdn.com/molmagb-170320021430/85/Magnetism-of-organometallics-with-DFT-an-alternate-approach-5-320.jpg)
![Ion substitution may be used to redesign MM
6
Cr8 Molecular Ring
Cr7Ni Molecular Ring
[1] M. Affronte et al., Chemical Communications, 1789 (2007).
[2] M. Affronte et al., Polyhedron 24, 2562 (2005).
[3] G. A. Timco et al., Nature Nanotechnology 4, 173 (2009).
[4] F. Troiani et al., Phys Rev Lett 94, 207208 (2005).](https://image.slidesharecdn.com/molmagb-170320021430/85/Magnetism-of-organometallics-with-DFT-an-alternate-approach-6-320.jpg)

![iiieff rV
)(
2
1 2
(1)
i
i rrn
2
)()( (2)
Kohn-Sham equations
][)()(
][][][)]([
nFdrrvrn
nVnVnTrnE
HKext
eeext
Hohenberg-Kohn functional
Electronic density n(r) determines all ground state
properties of multi-electron system. Energy of the
ground state is a functional of electronic density:
Density Functional Theory (DFT)
prediction of J from first principles
8
Where are KS orbitals, is the system of N effective one-particle equations](https://image.slidesharecdn.com/molmagb-170320021430/85/Magnetism-of-organometallics-with-DFT-an-alternate-approach-8-320.jpg)
![Energy can be predicted
for high and low spin states
9
Density Functional Theory (DFT)
E=E[ρ]
to simplify Kinetic part, total electron density is separated
into KS orbitals, describing 1e each:
Electron interaction accounted for self-consistently via
exchange-correlation potential
)()()'
|'|
)'(
( 2
2
1
rrVdr
rr
r
V iiixcext
2
1
|)(|)( rr i
N
i
i
](https://image.slidesharecdn.com/molmagb-170320021430/85/Magnetism-of-organometallics-with-DFT-an-alternate-approach-9-320.jpg)

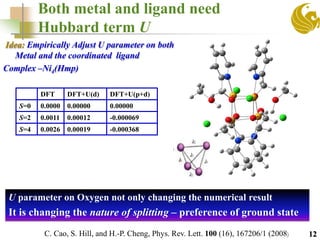
![Numeric values of U parameters for
different atom types are fitted using
benchmark set
Chemical formula
J (cm-1)
Plane Wave
calculations
BS-DFT Expt
DFT+U
metal+ligand
DFT+U
metal only
[Mn2 (IV)(μO)2 (phen)4]4+ -143.6 -166.6 -131.9 -147.0
[Mn2(IV)(μO)2((ac))(Me4dtne)]3+ -74.9 -87.4 -37.5 -100.0
[Mn2(III) (μO)(ac)2(tacn)2]2+ 5.6 -3.64 -40.0 10.0
[Mn2(II) (ac)3(bpea)2]+ -7.7 -18.8 - -1.3
[Mn(III)Mn(IV)(μO)2(ac)(tacn)2]2+ -234.0 -247.6 -405 -220
13
U (Mn)=2.1 eV, U(O)=1.0 eV, U(N)=0.2 eV](https://image.slidesharecdn.com/molmagb-170320021430/85/Magnetism-of-organometallics-with-DFT-an-alternate-approach-13-320.jpg)
![(Mn(IV))2 (OAc)
Exp BSDFT DFT+U
-100 -37 -74.9
Computational Details
Cutoff
25 Ryd
Smearing
Marzari-Vanderbilt cold smearing
Smearing Factor
0.0008
For better convergence
Local Thomas Fermi screening
14
Evaluation of J(cm-1)
We modify the source code of Quantum ESPRESSO to incorporate
U on Nitrogen
[Mn2(IV)(μO)2((ac))(Me4dtne)]3+](https://image.slidesharecdn.com/molmagb-170320021430/85/Magnetism-of-organometallics-with-DFT-an-alternate-approach-14-320.jpg)
![Mn(IV)- no acetate bridge
Exp BSDFT DFT+U
-147 -131 -164
15
Evaluation of J(cm-1)
[Mn2 (IV)(μO)2 (phen)4]4+](https://image.slidesharecdn.com/molmagb-170320021430/85/Magnetism-of-organometallics-with-DFT-an-alternate-approach-15-320.jpg)
![Exp BSDFT DFT+U
10 -40 29
16
Mn(III)
two acetate bridges
Evaluation of J(cm-1)
Exp BSDFT DFT+U
-1.5 -8
Mn(II)
three acetate bridges
[Mn2(II) (ac)3(bpea)2]+
[Mn2(III) (μO)(ac)2(tacn)2]2+](https://image.slidesharecdn.com/molmagb-170320021430/85/Magnetism-of-organometallics-with-DFT-an-alternate-approach-16-320.jpg)
![17
J cm-1 (MnIII-MnIV)
Exp BSDFT DFT+U
-220 -155 -234
Mixed valence Mn(III)-Mn(IV)
[Mn(III)Mn(IV)(μO)2(ac)(tacn)2]2+](https://image.slidesharecdn.com/molmagb-170320021430/85/Magnetism-of-organometallics-with-DFT-an-alternate-approach-17-320.jpg)

![Failure of BSDFT
Bimetallic complexes with Acetate Bridging ligand
Complexes with Ferromagnetic Coupling
Mix valence complexes
20
Chemical formula
J (cm-1)
Plane Wave
calculations
BS-DFT Expt
DFT+U
metal+ligand
DFT+U
metal only
[Mn2(IV)(μO)2((ac))(Me4dtne)]3+ -74.9 -87.4 -37.5 -100.0
[Mn2(III) (μO)(ac)2(tacn)2]2+ 5.6 -3.64 -40.0 10.0
[Mn(III)Mn(IV)(μO)2(ac)(tacn)2]2+ -234.0 -247.6 -405 -220](https://image.slidesharecdn.com/molmagb-170320021430/85/Magnetism-of-organometallics-with-DFT-an-alternate-approach-20-320.jpg)
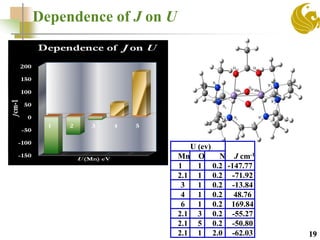
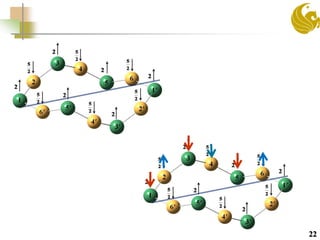
![Two qubit system-[Mn12(Reda)] complex with
weakly coupled subsystems
21
Predict J for two
coupled sub system
Previous DFT Study predicted J=0
Whereas the J>0 experimentally
Methyl diethanolamine Allyl diethanolamine](https://image.slidesharecdn.com/molmagb-170320021430/85/Magnetism-of-organometallics-with-DFT-an-alternate-approach-21-320.jpg)
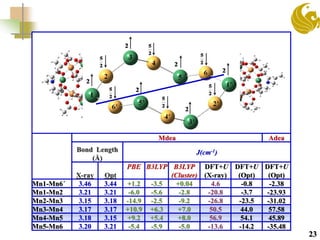
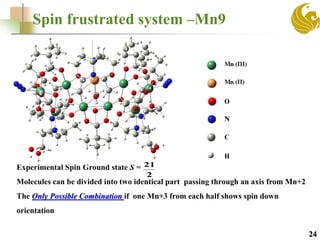
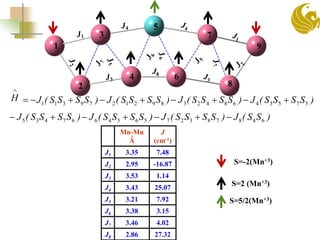
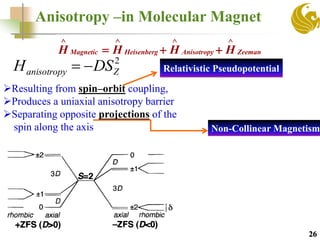
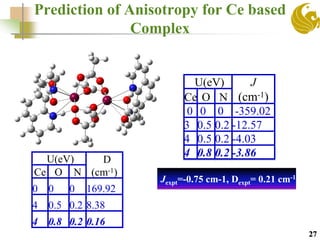




The document discusses the use of first principle calculations and the DFT+U method to predict magnetic coupling in organometallic complexes, particularly in molecular magnets that may serve as qubits in quantum computing. Key topics include benchmarking studies of various two-qubit systems, spin frustration, and the need for accurate predictions of magnetic properties through reliable approaches. Future research plans focus on improving computational methods to refine magnetic property predictions and exploring the anisotropy of molecular magnets.



































































