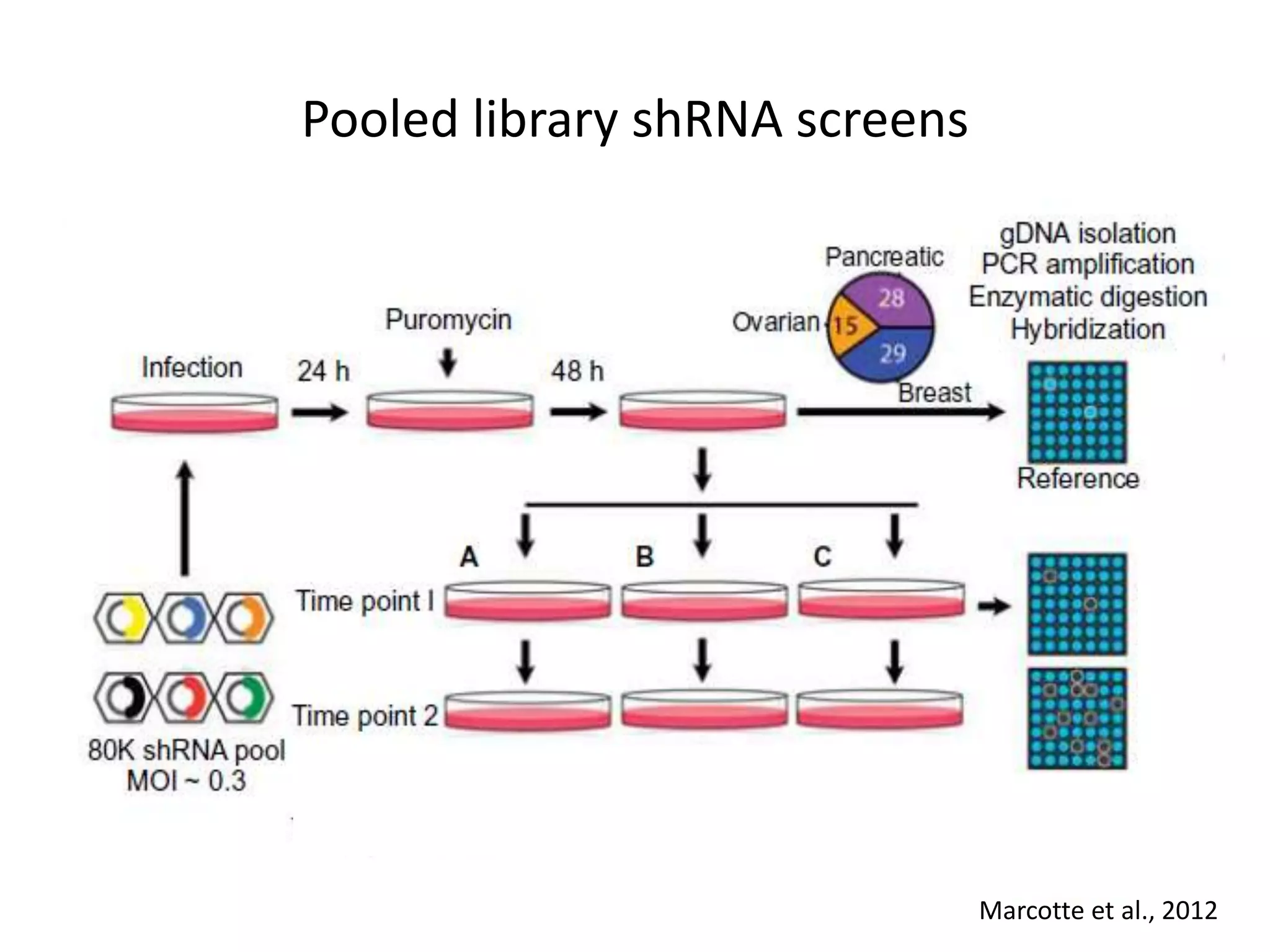
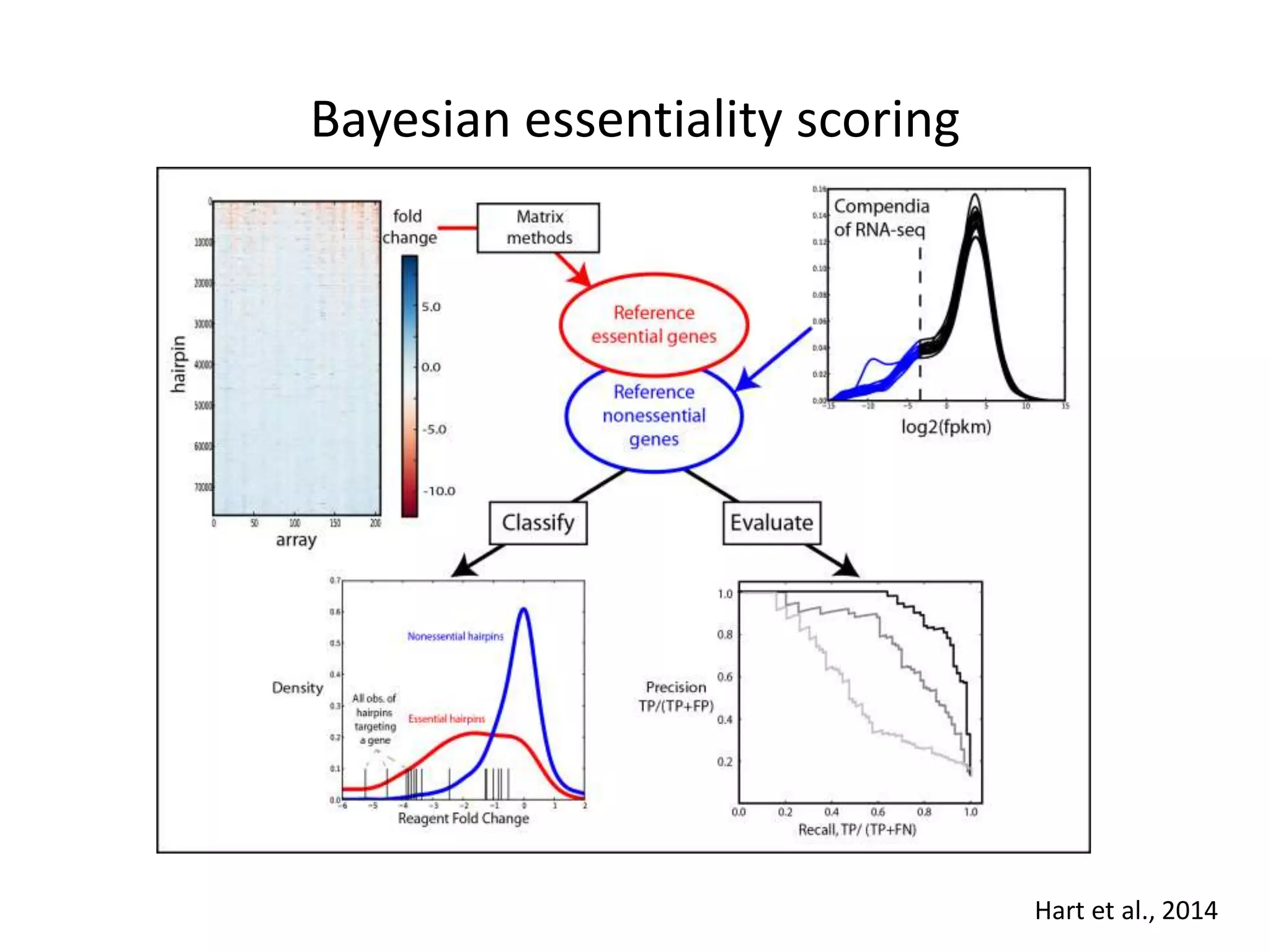
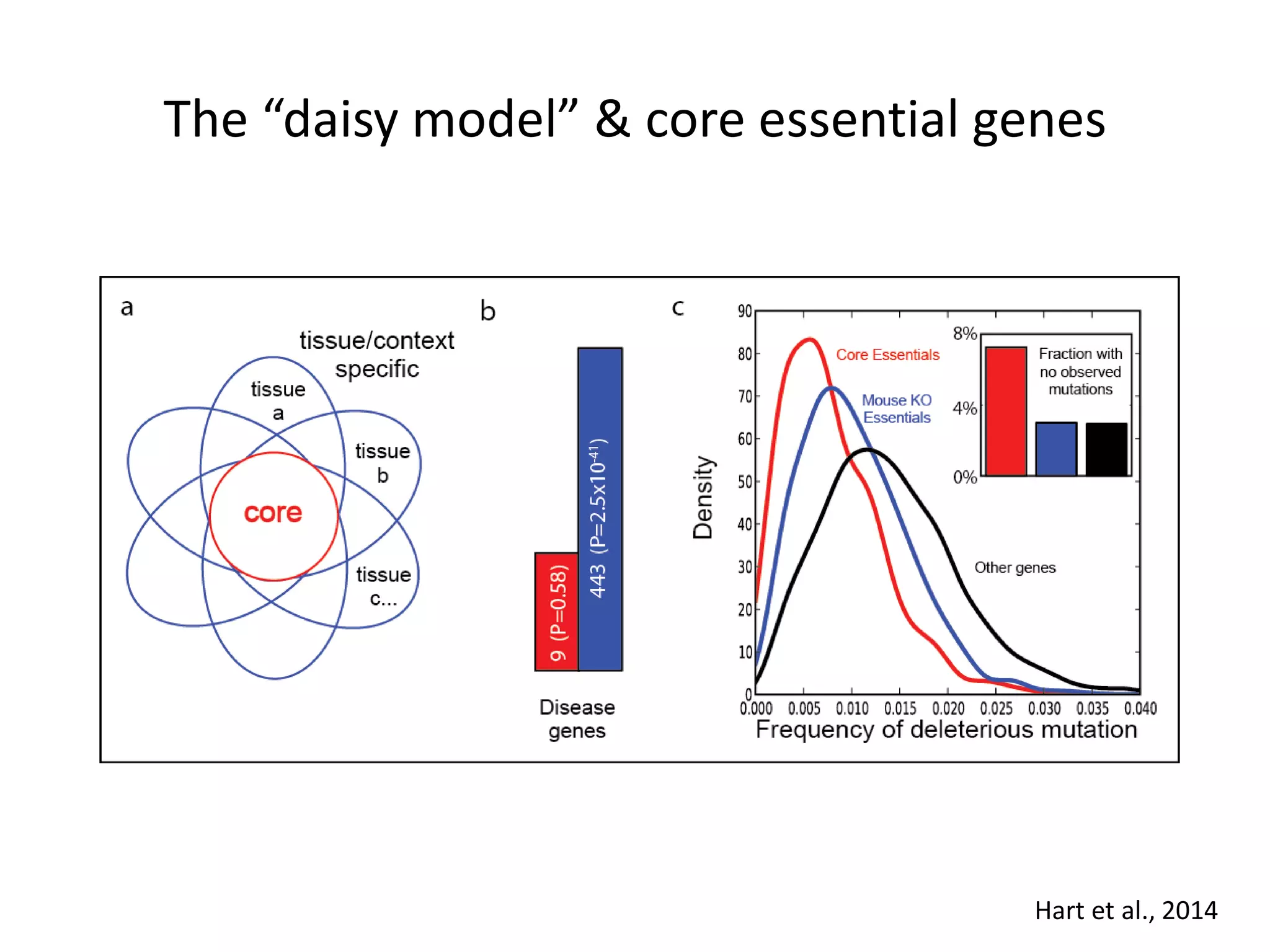
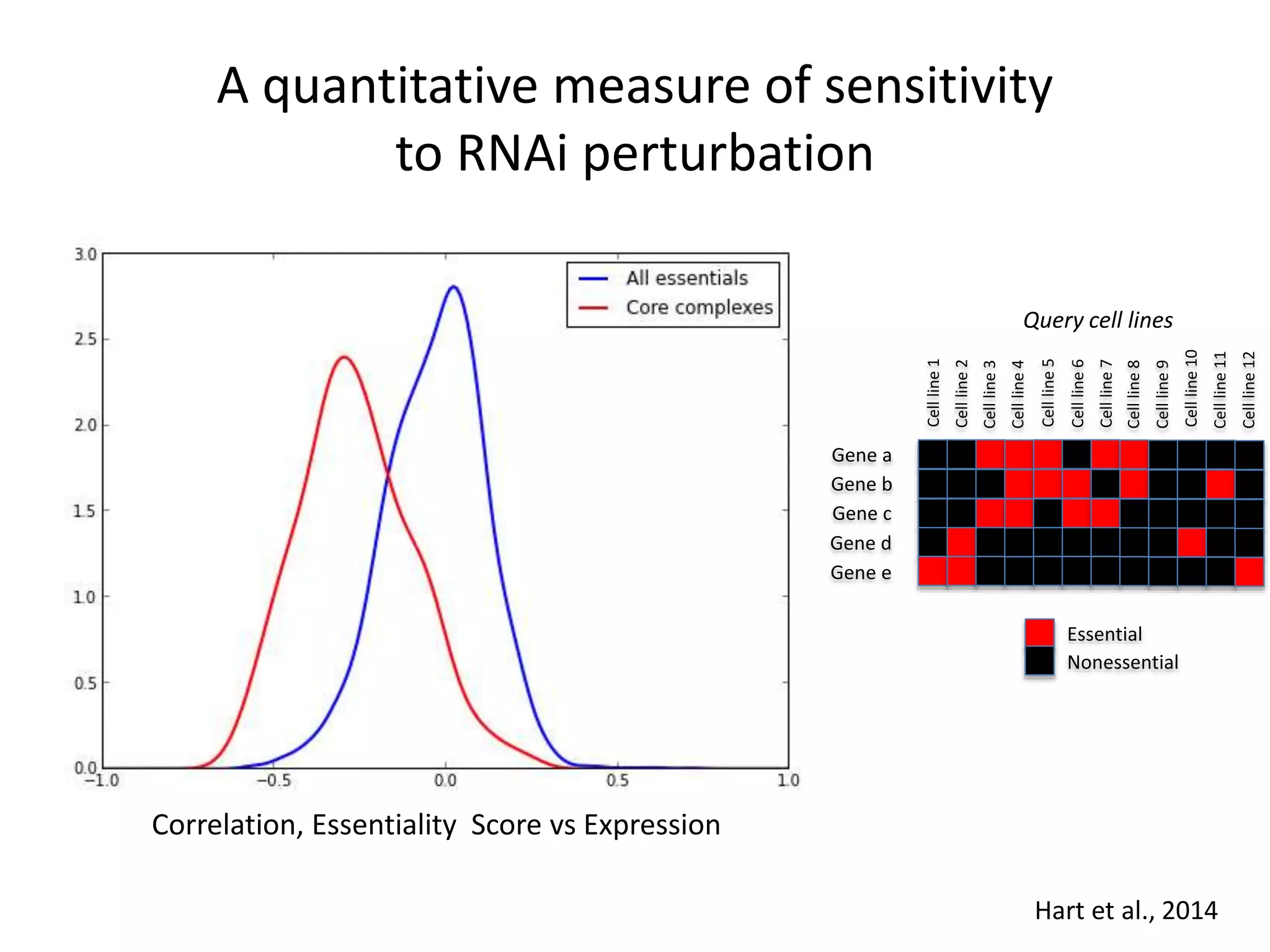
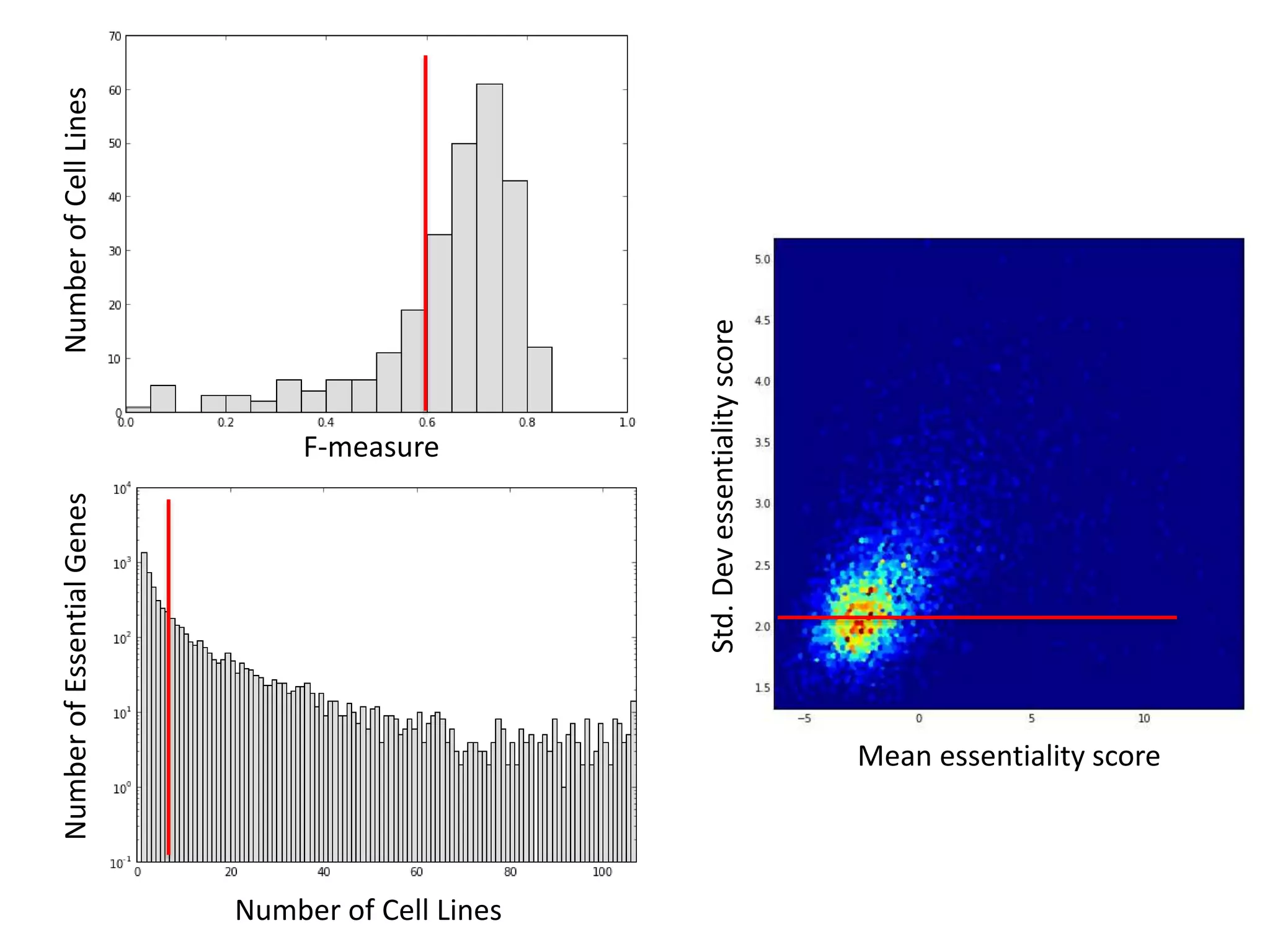
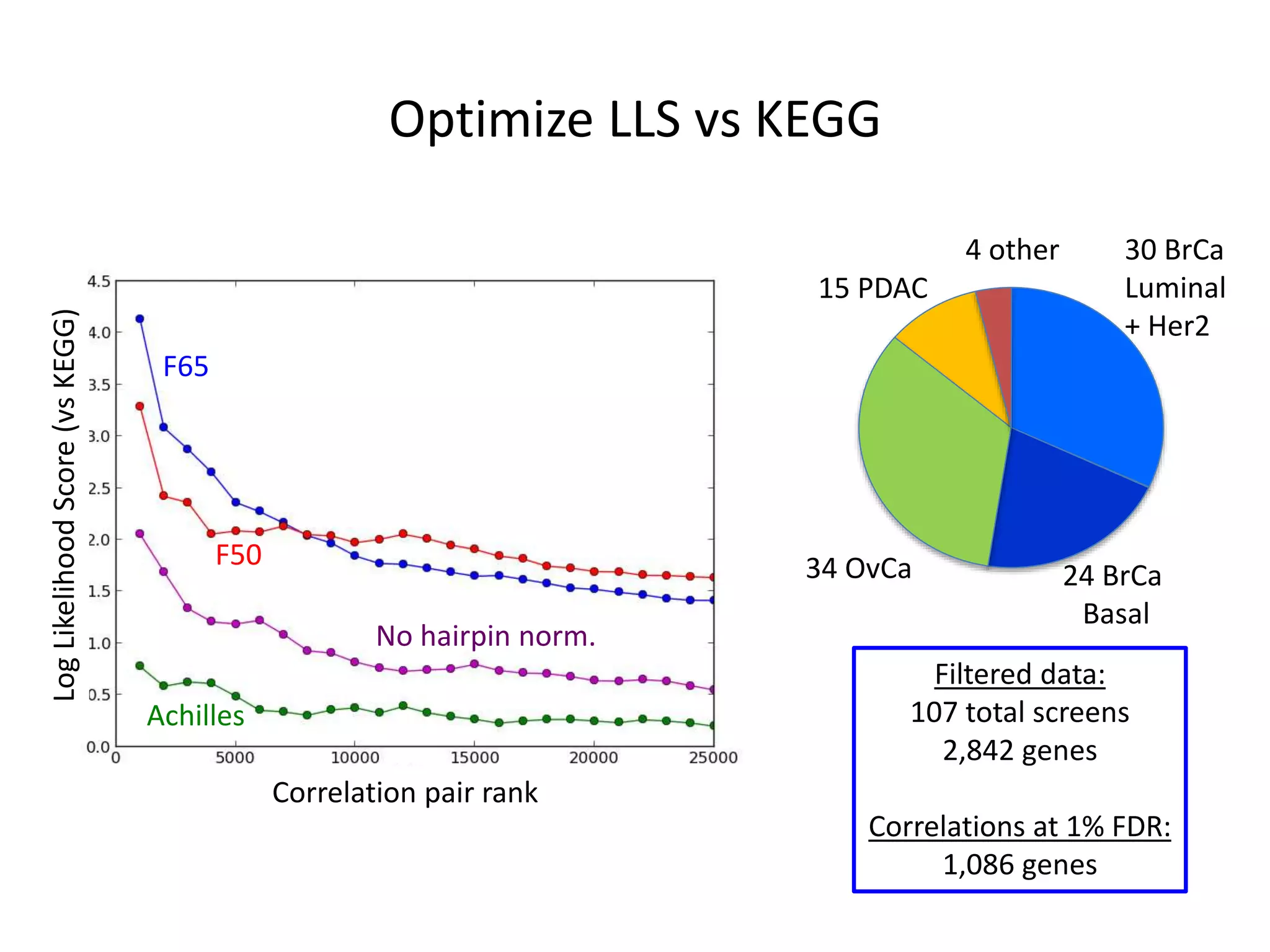
This document summarizes research on developing a human cancer coessentiality network using data from pooled shRNA screens across 107 cancer cell lines. Key points:
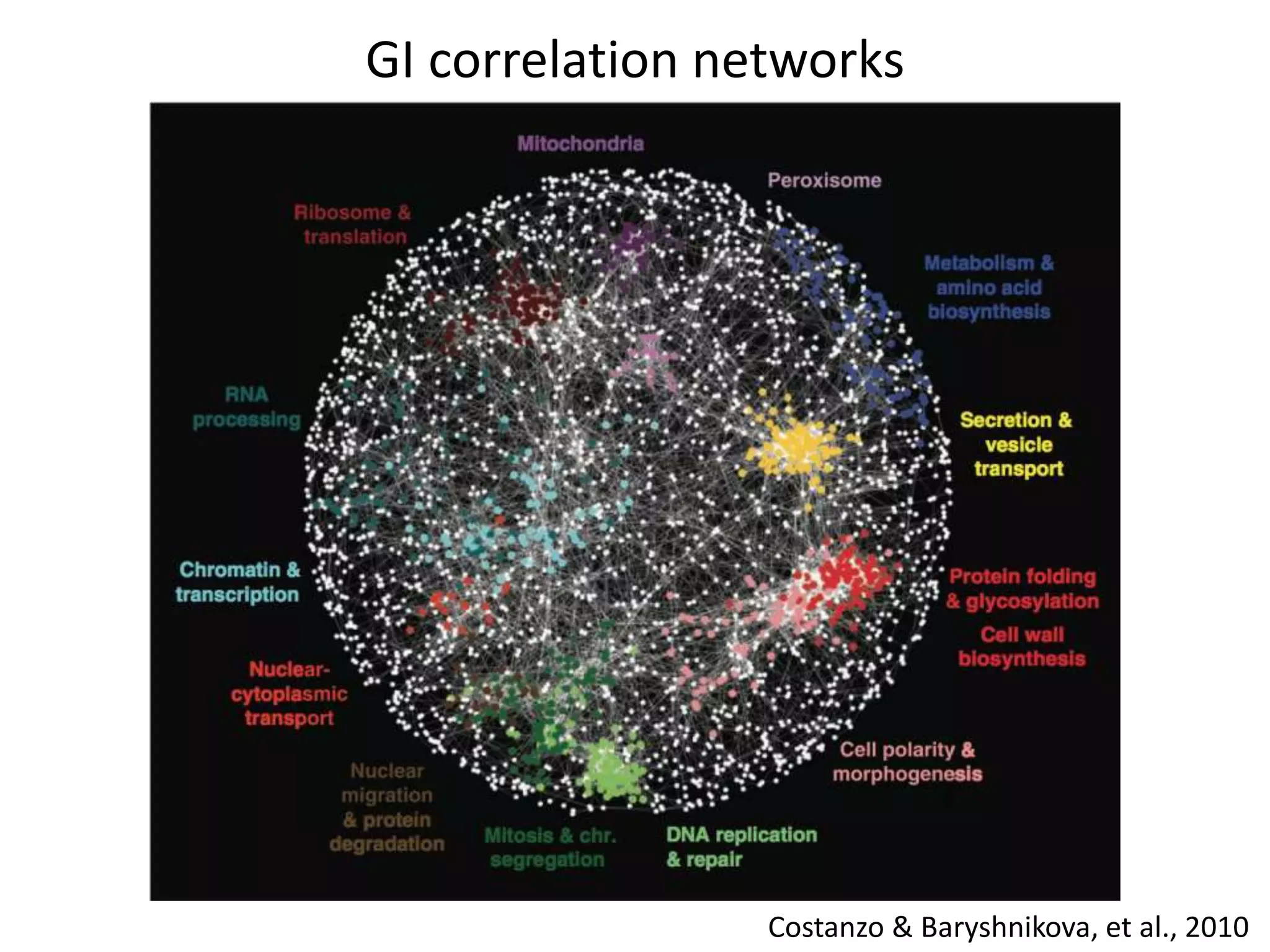
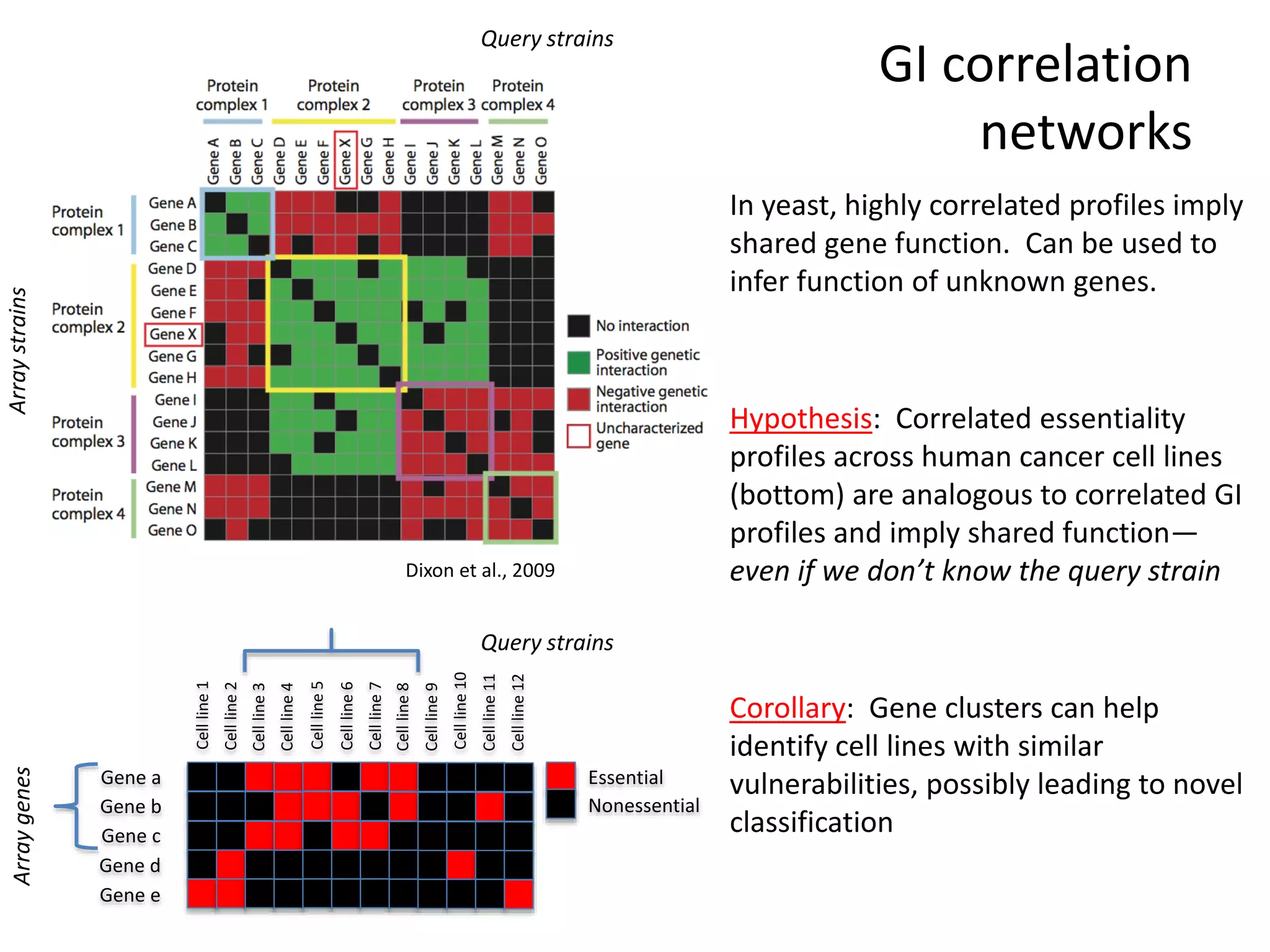
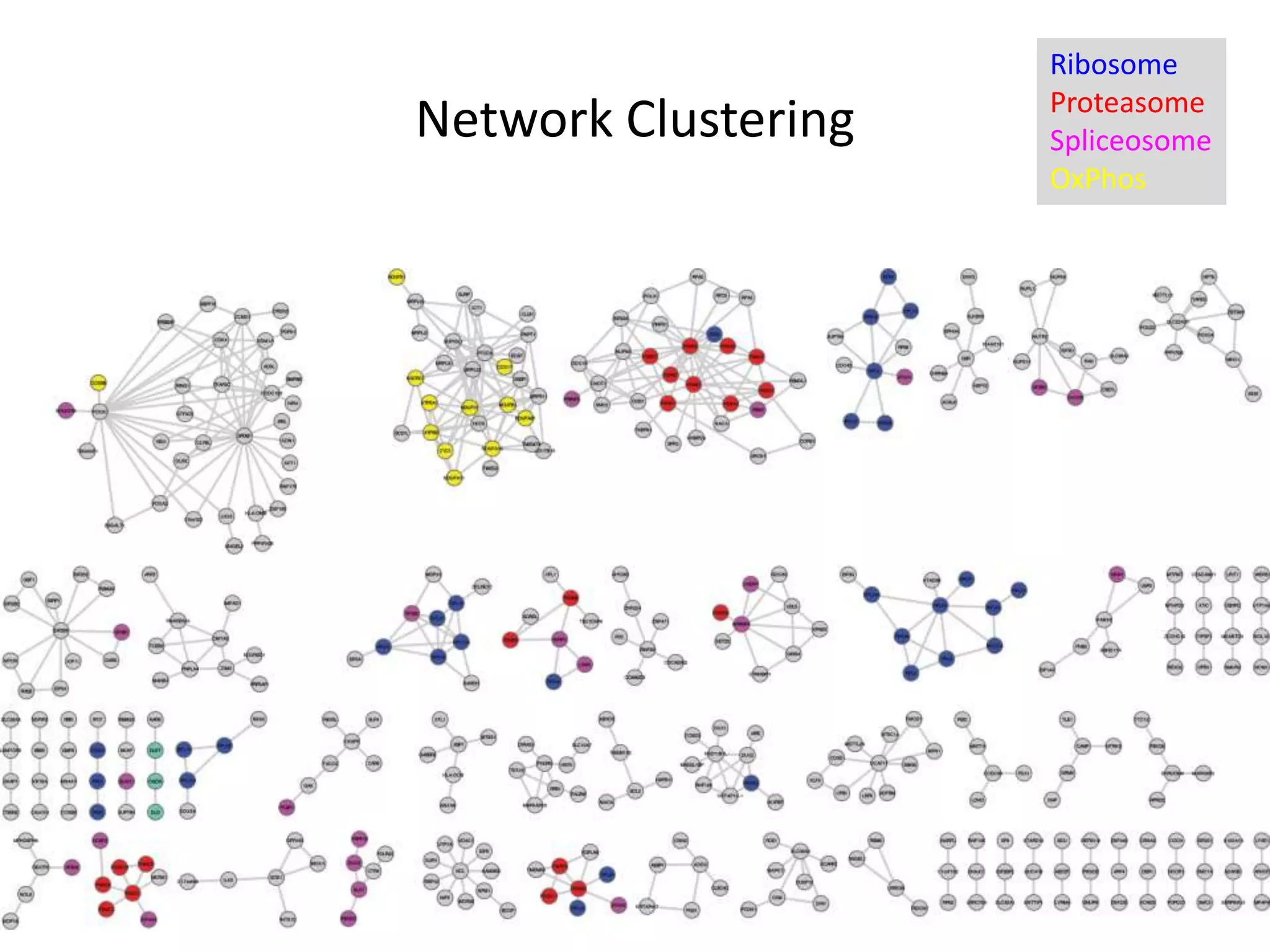
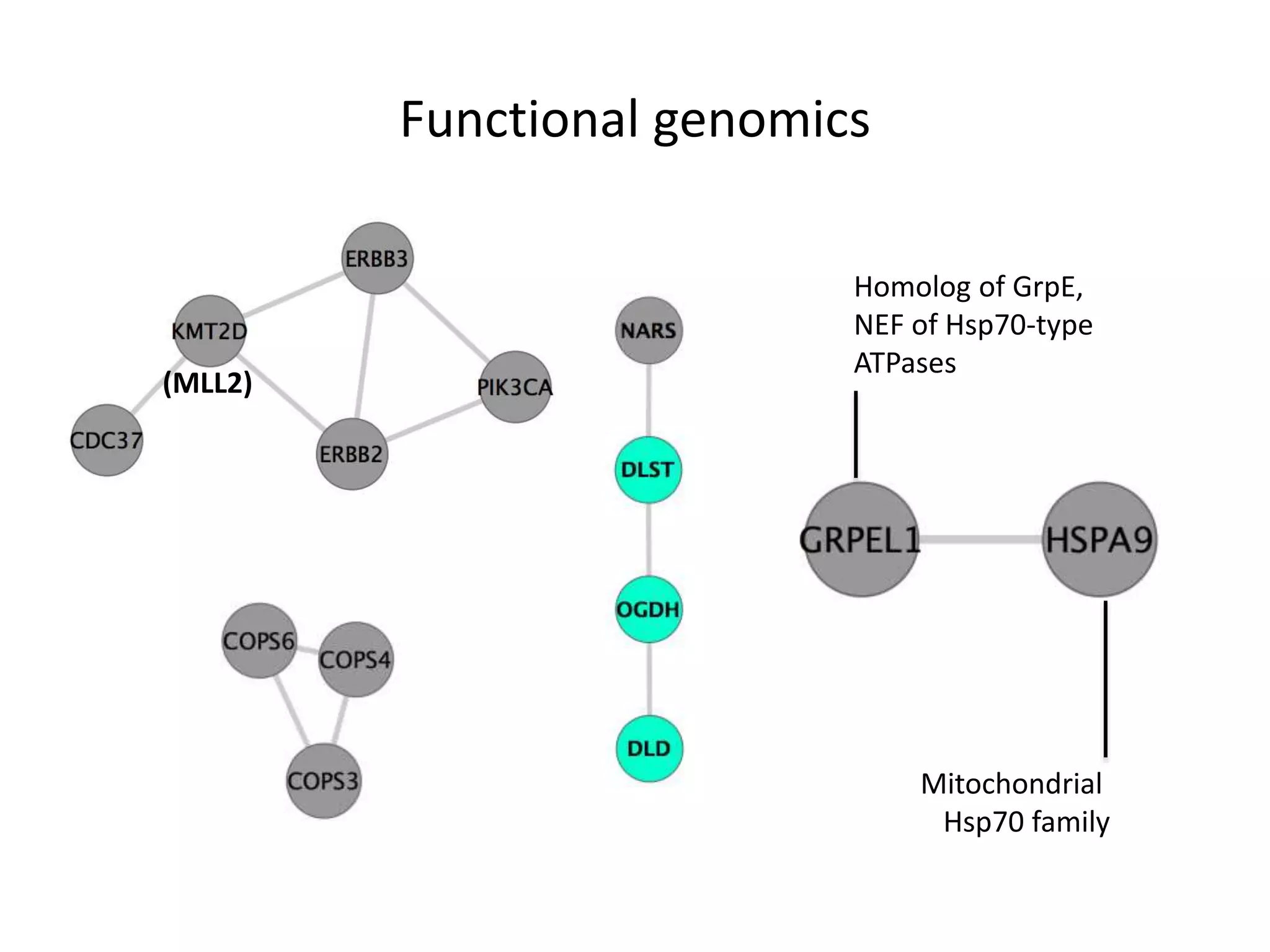
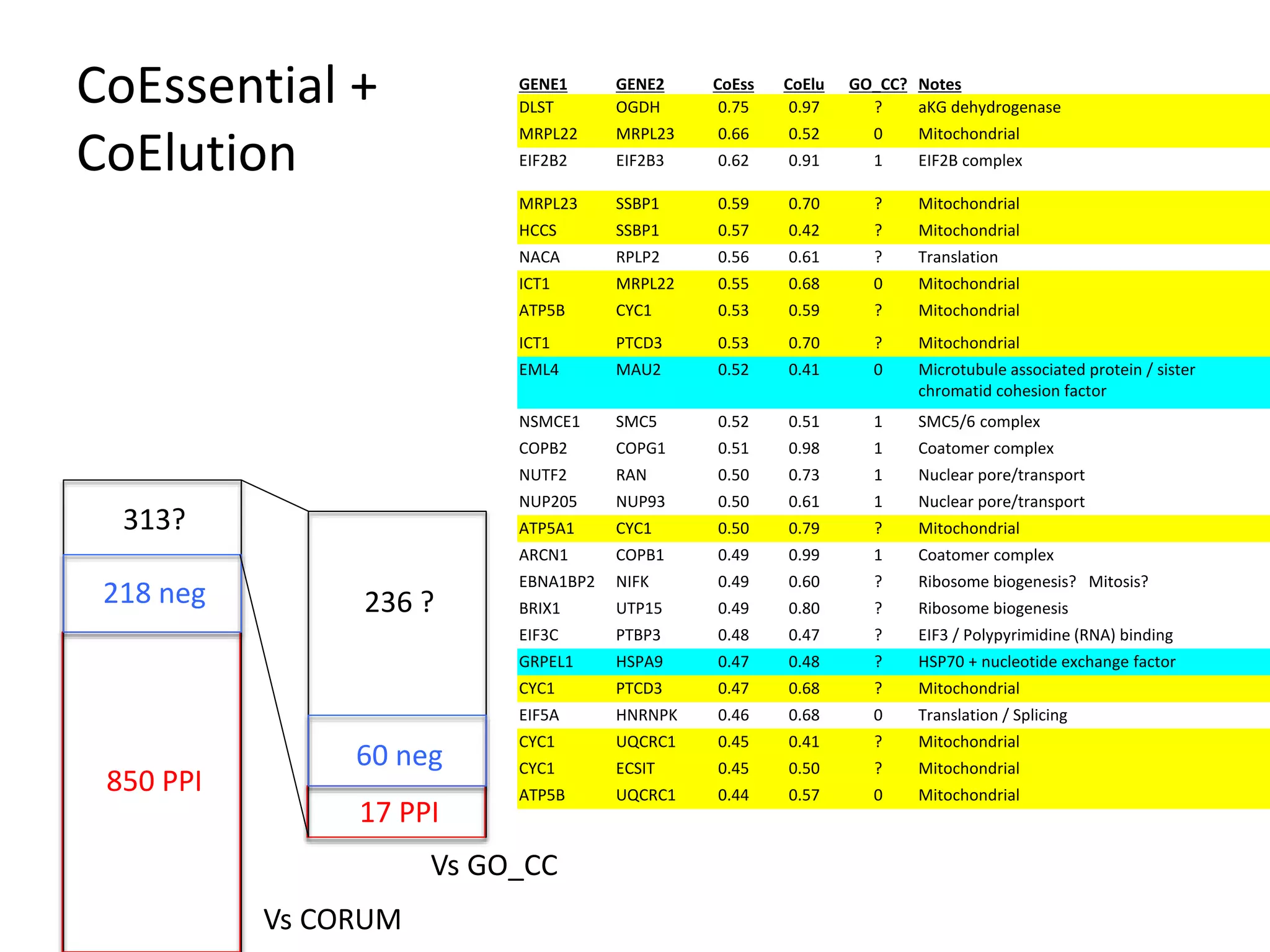
- A network of 866 genes and 1877 edges was constructed based on correlations in essentiality profiles across cell lines.
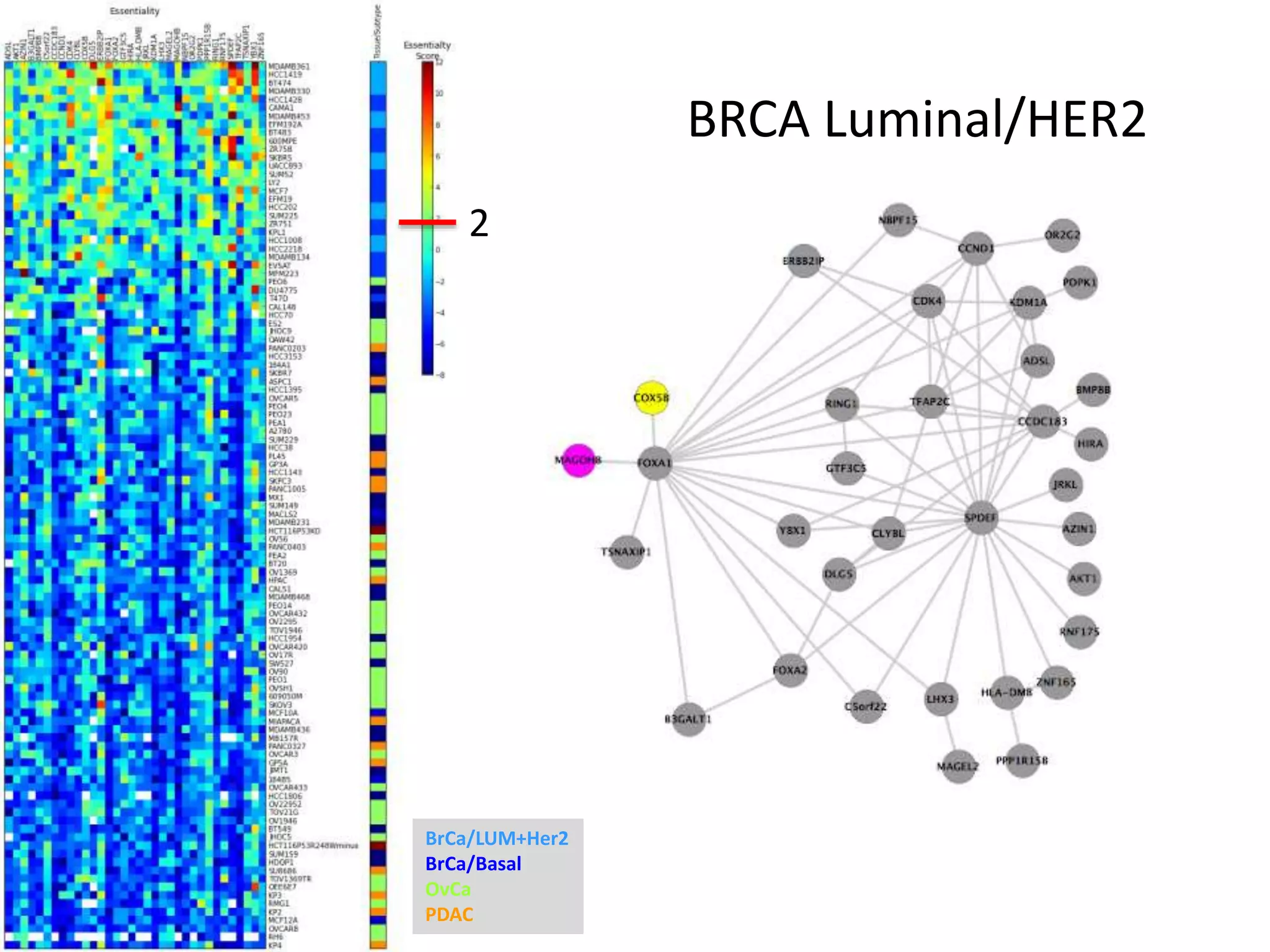
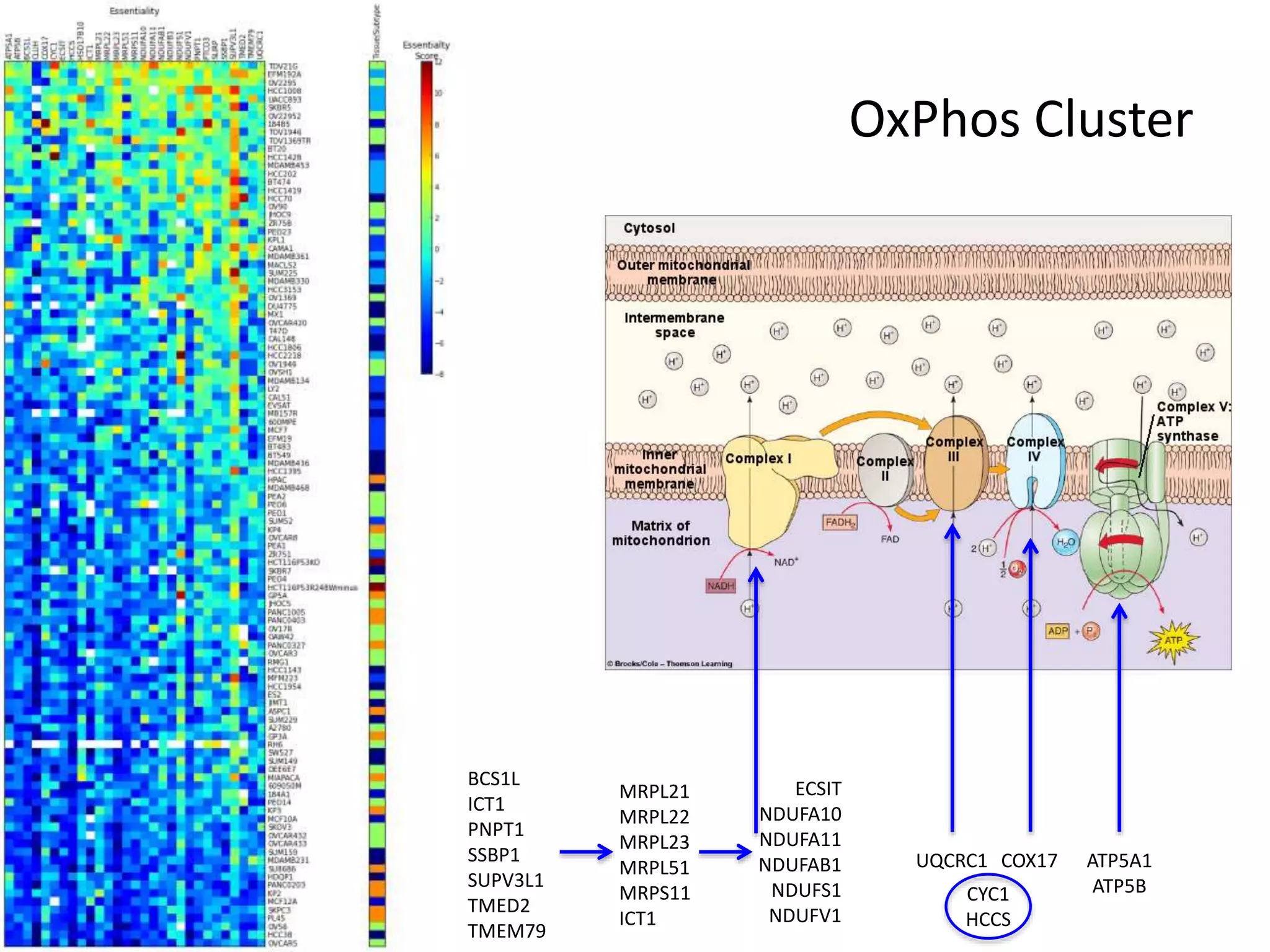
- Network clustering identified groups of genes essential for similar cell line subtypes (e.g. breast, ovarian, pancreatic cancers).
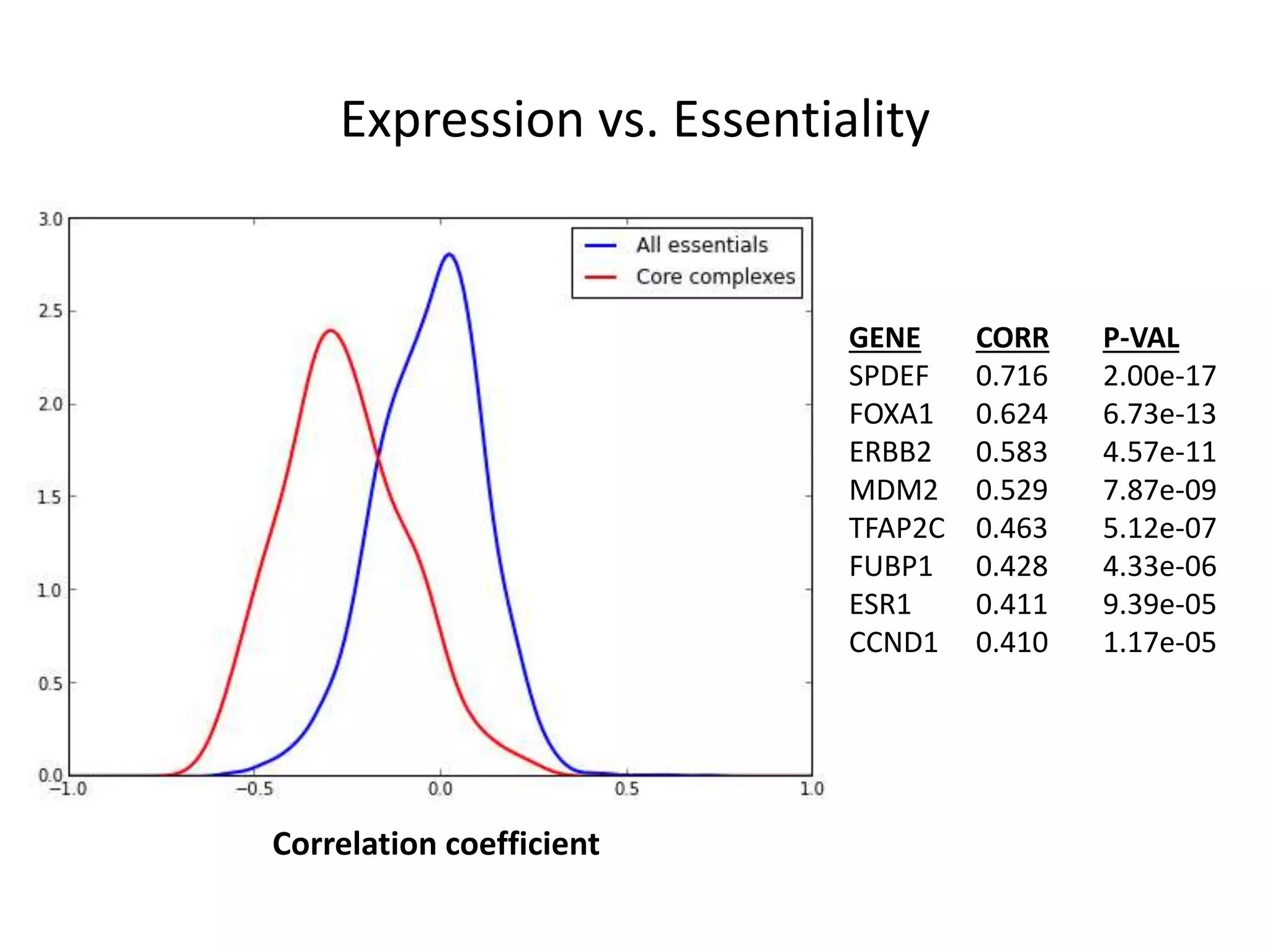
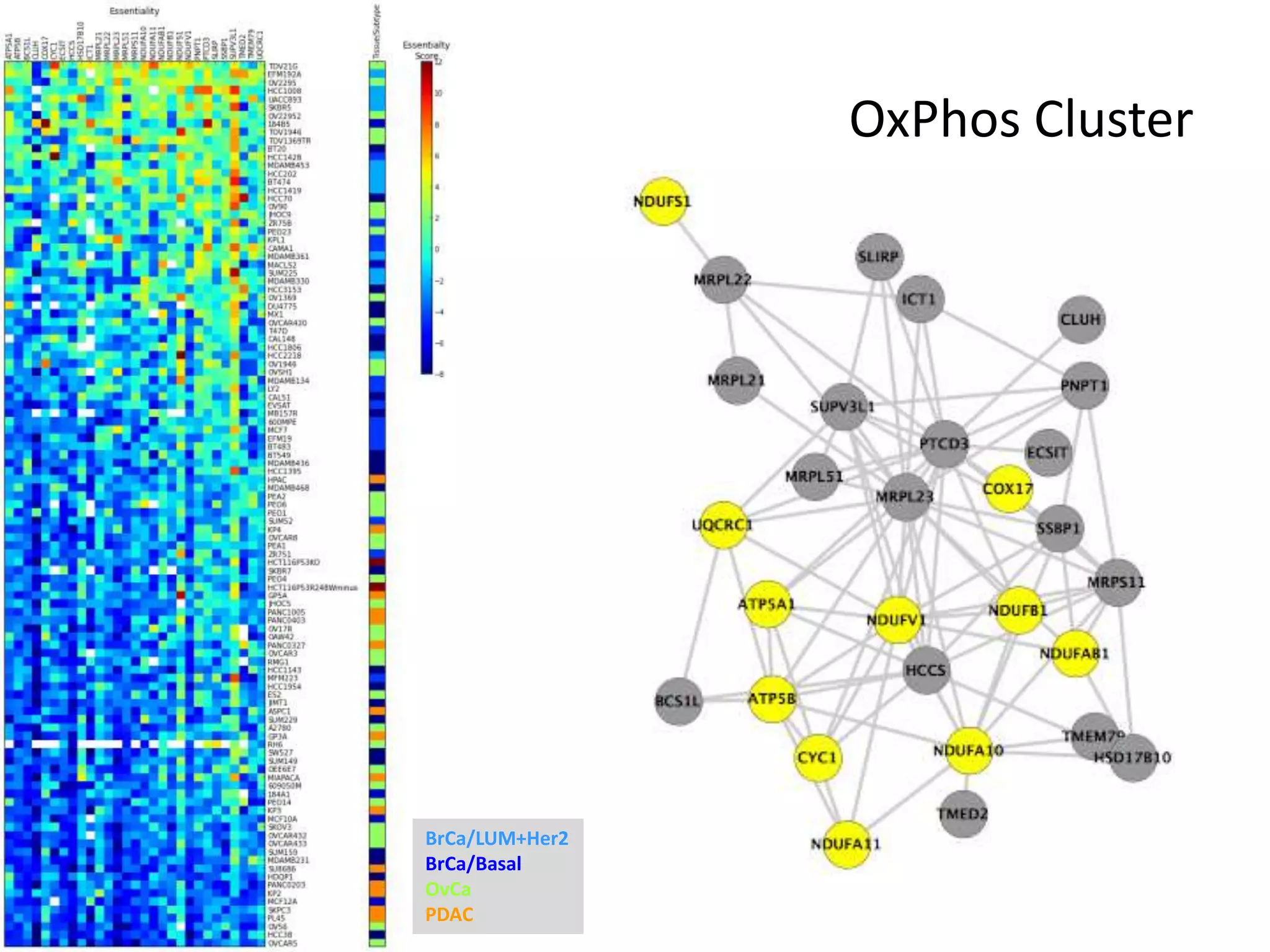
- One cluster involved in oxidative phosphorylation was particularly essential for luminal/HER2 breast cancers.
- The network provides a functional genomics resource, though opportunities exist to improve coverage and accuracy.