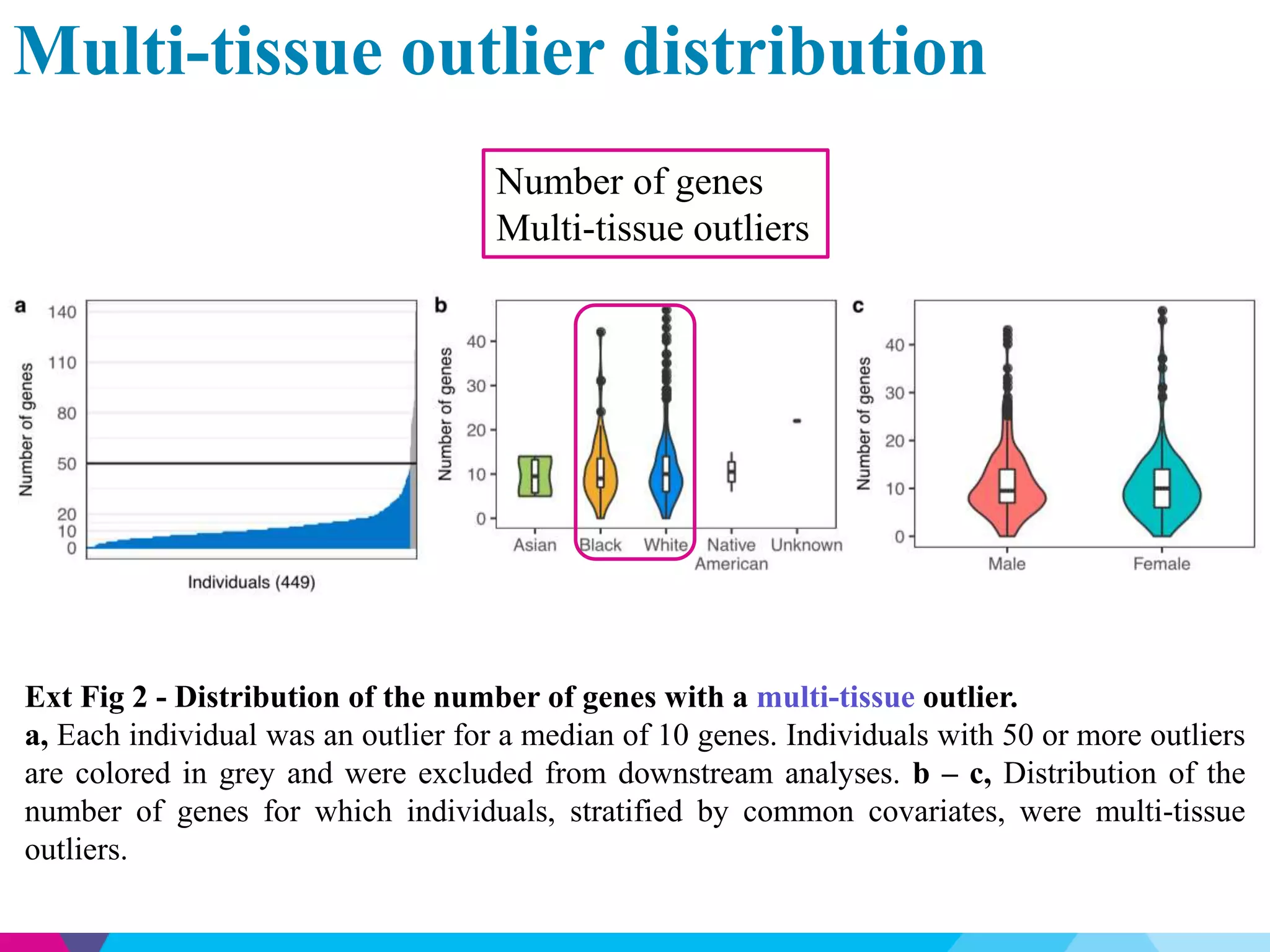
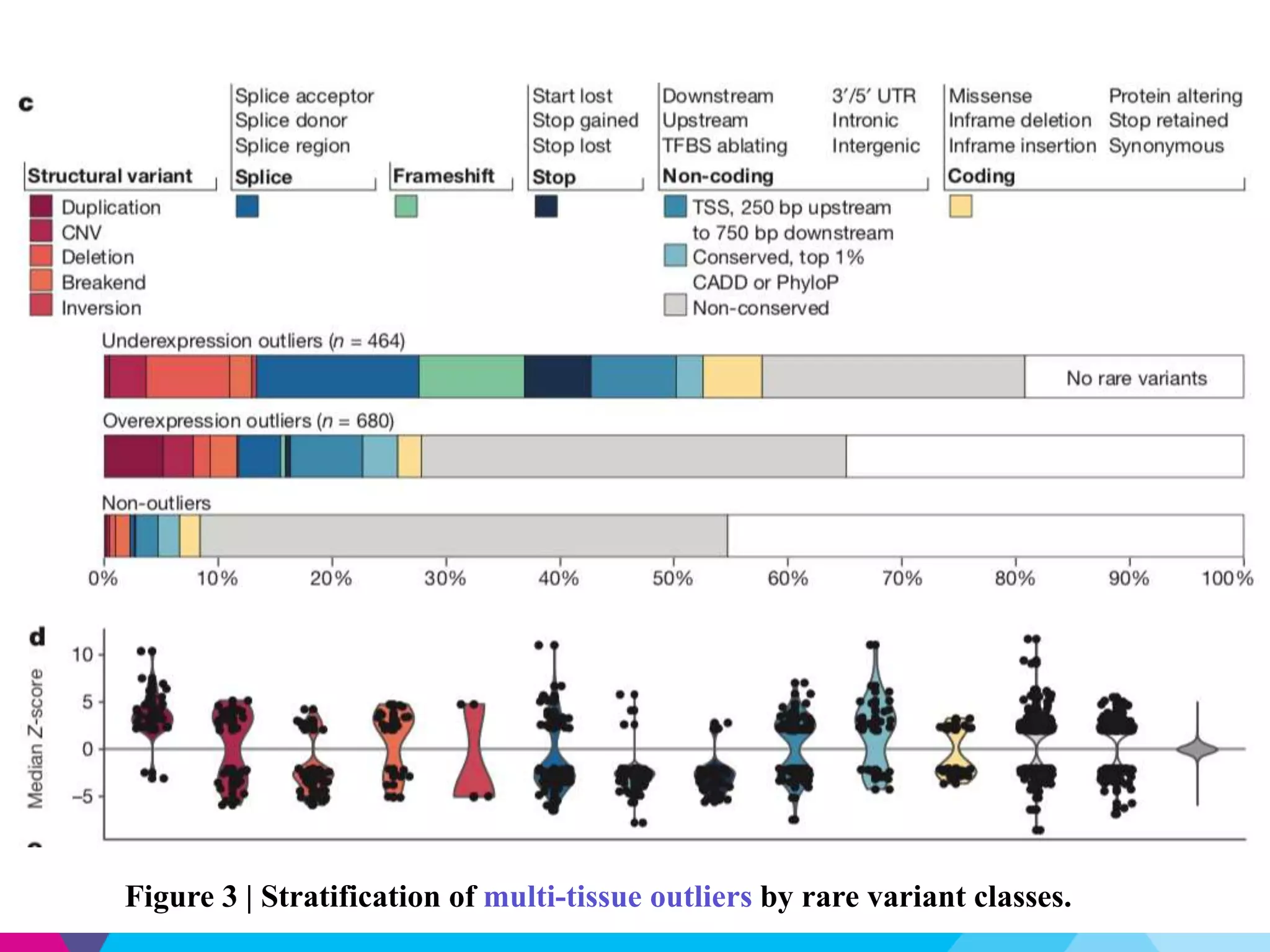
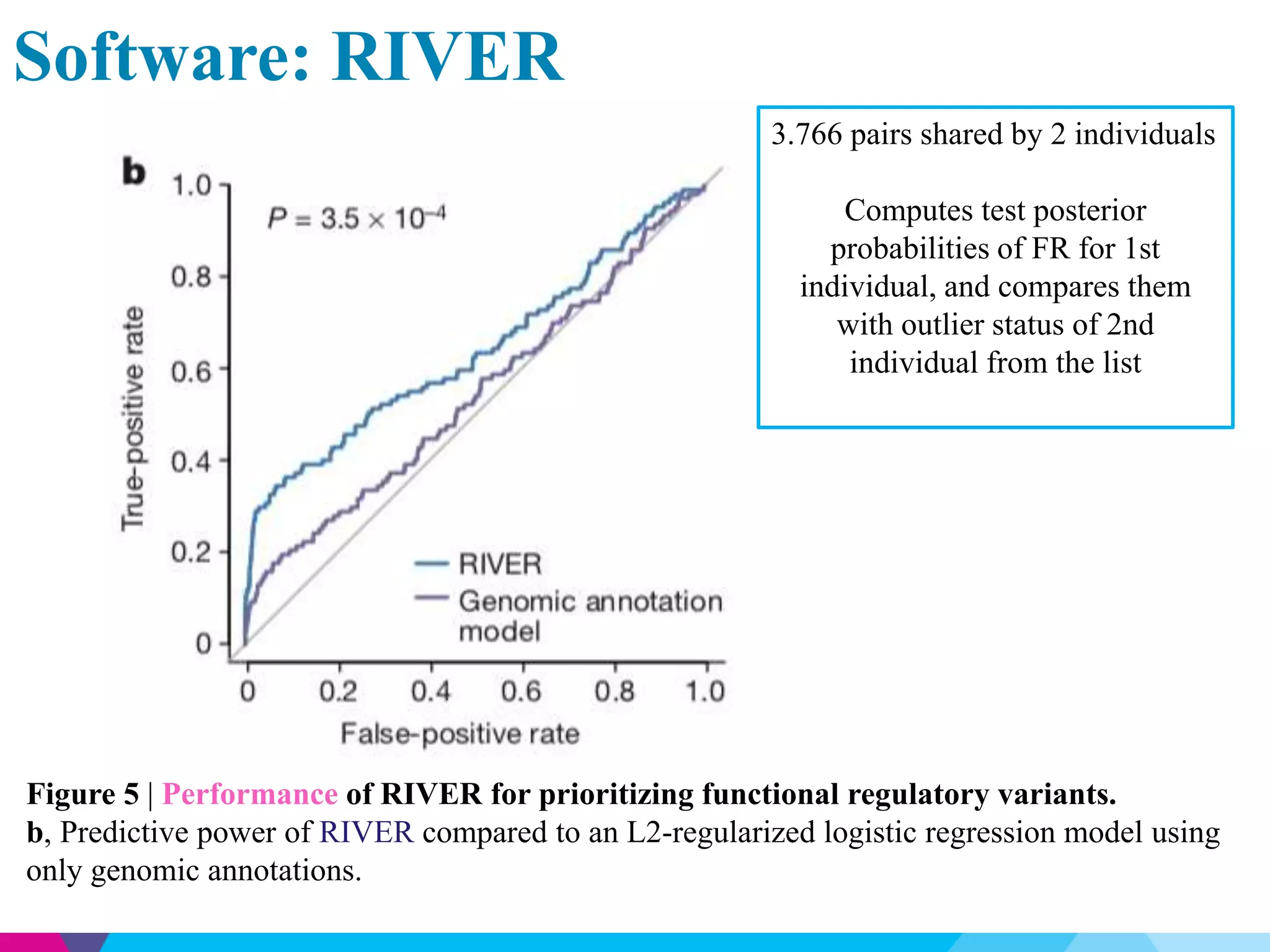
This document discusses the impact of rare genetic variations on gene expression across different tissues, highlighting the challenges in distinguishing pathogenic from benign variants and the inefficacy of traditional association studies for rare variants. Utilizing data from the GTEx project, the analysis reveals that multi-tissue expression outliers are significantly enriched for nearby rare variants, particularly structural variants and indels. The study also introduces a Bayesian model, RIVER, to assess the regulatory effects of these variants on gene expression, demonstrating its predictive power compared to traditional models.